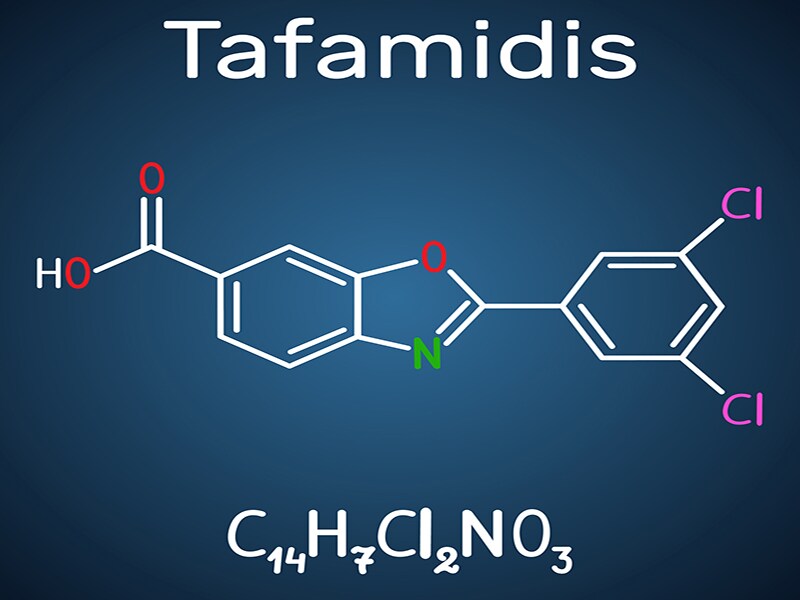
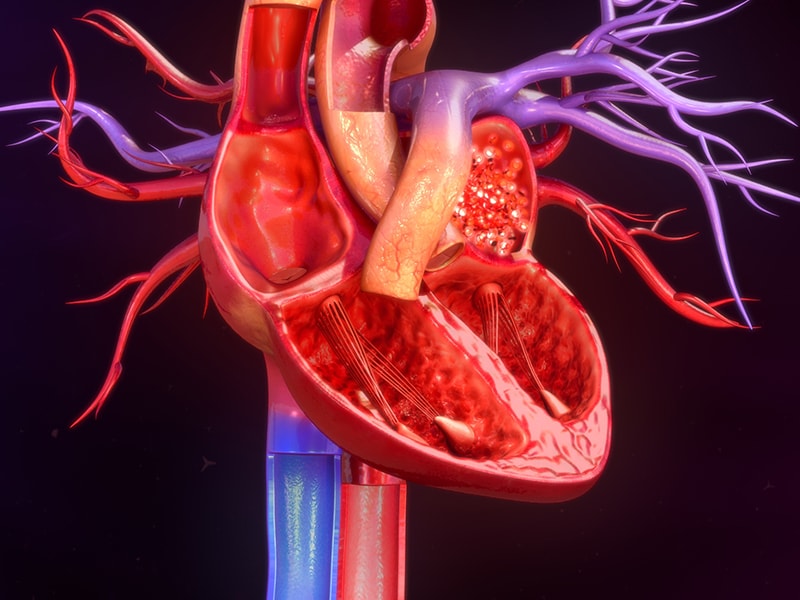
Amyloïdose
Amyloïde
Protéine Amyloïde A Sérique
Préalbumine
Chaînes Légères Des Immunoglobulines
Fièvre Méditerranéenne Familiale
Neuropathies Amyloïdes
Rouge Congo
Neuropathies Amyloïdes Familiales
Cardiomyopathies
Bêta-2-Microglobuline
Composant Amyloïde P Sérique
Syndrome Néphrotique
Paraprotéinémies
Maladie De La Trachée
Macroglossie
Amyloidogenic Proteins
Melphalan
Protéine Bence Jones
Biopsie
Acinonyx
Chaînes Légères Lambda Des Immunoglobulines
Gelsoline
Déficit En Facteur X
Maladies Génétiques De La Peau
Fatal Outcome
Myélome Multiple
Troponine
Troponine C
Troponine T
Encyclopedias as Topic
Myosines
Troponine I
L'amylose est une maladie rare mais grave qui se caractérise par l'accumulation anormale et généralisée d'une protéine appelée "fibrilles amyloïdes" dans différents tissus et organes du corps. Ces fibrilles sont des agrégats de protéines mal repliées qui forment des dépôts insolubles et résistants, perturbant ainsi la structure et la fonction normales des organes touchés.
L'amylose peut affecter divers systèmes corporels, tels que le cœur, les reins, le foie, les glandes surrénales, les poumons, le système nerveux périphérique et la peau. Les symptômes varient considérablement en fonction des organes atteints et peuvent inclure :
* Insuffisance cardiaque congestive
* Insuffisance rénale
* Neuropathie sensorielle ou motrice
* Hépatomégalie (augmentation du volume du foie)
* Splénomégalie (augmentation du volume de la rate)
* Purpura cutané (ecchymoses et petites hémorragies sous-cutanées)
Il existe plusieurs types d'amylose, classés en fonction de la protéine spécifique qui forme les dépôts amyloïdes. Les plus courants sont l'amylose AL (associée à une production anormale de chaînes légères d'immunoglobulines par des cellules plasmocytaires malignes ou bénignes), l'amylose AA (liée à une inflammation chronique) et l'amylose héréditaire ou familiale (due à des mutations génétiques spécifiques).
Le diagnostic d'amylose repose sur des tests de laboratoire, des examens d'imagerie et des biopsies tissulaires pour confirmer la présence de dépôts amyloïdes. Le traitement dépend du type d'amylose et peut inclure une chimiothérapie, une greffe de cellules souches, des médicaments anti-inflammatoires ou des thérapies ciblant la protéine responsable de la formation des dépôts amyloïdes.
L'amyloidose familiale est une maladie génétique rare caractérisée par l'accumulation anormale et tissulaire de protéines amyloïdes, ce qui peut affecter différents organes du corps. Il existe plusieurs types d'amyloidose familiale, chacun étant lié à des mutations spécifiques dans certains gènes. Les symptômes et l'évolution de la maladie peuvent varier considérablement en fonction du type et de la localisation des dépôts amyloïdes.
Les protéines amyloïdes sont des molécules anormalement pliées qui ont tendance à s'agréger et à former des dépôts dans les tissus. Dans l'amyloidose familiale, ces dépôts sont héréditaires en raison de mutations génétiques spécifiques. Les protéines amyloïdes les plus souvent touchées dans cette maladie sont la transthyrétine (TTR) et l'apolipoprotéine A-I (APOA1).
Les symptômes de l'amyloidose familiale peuvent inclure une neuropathie périphérique, des troubles cardiaques, des insuffisances rénales, des problèmes gastro-intestinaux et une hépatomégalie (augmentation du volume du foie). La présentation clinique peut varier considérablement d'un type à l'autre.
Le diagnostic de l'amyloidose familiale repose généralement sur des tests génétiques pour détecter les mutations spécifiques associées à la maladie, ainsi que sur des biopsies tissulaires pour confirmer la présence de dépôts amyloïdes. Le traitement vise principalement à ralentir ou à arrêter la progression de la maladie et peut inclure des médicaments pour stabiliser les protéines amyloïdes, des thérapies visant à réduire la production de ces protéines et des transplantations d'organes dans certains cas.
Il est important de noter que l'amyloidose familiale est une maladie héréditaire, ce qui signifie qu'elle peut être transmise de génération en génération. Les personnes ayant des antécédents familiaux de cette maladie ou présentant des symptômes évocateurs devraient consulter un médecin pour un diagnostic et une prise en charge appropriés.
L'amyloïde est une protéine anormalement pliée et accumulée qui se dépose sous forme de fibrilles bêta-pliées dans les tissus et organes, entraînant leur dysfonctionnement. Ces dépôts amyloïdes peuvent s'accumuler dans divers endroits du corps, mais ils sont souvent trouvés dans le cerveau, le cœur, les reins, le foie et les glandes surrénales.
Dans le cerveau, l'amyloïde est associée à la maladie d'Alzheimer et à d'autres troubles neurodégénératifs. Les plaques amyloïdes forment des dépôts extracellulaires entre les neurones, perturbant ainsi la communication neuronale et entraînant une toxicité cellulaire.
Dans le cœur, l'amyloïde peut provoquer une cardiomyopathie restrictive, une insuffisance cardiaque et des arythmies. Dans les reins, le foie et les glandes surrénales, l'amyloïde peut entraîner une insuffisance organique en altérant la structure et la fonction des tissus affectés.
Il existe différents types d'amyloïdes, chacun étant associé à une protéine spécifique qui se replie maladroitement. Par exemple, l'amyloïde-β est le type principal trouvé dans la maladie d'Alzheimer, tandis que l'amyloïde A est souvent observée dans les maladies inflammatoires et infectieuses chroniques.
Le diagnostic de l'amyloïose, qui est le terme utilisé pour décrire la présence généralisée d'amyloïdes dans le corps, repose sur des tests de laboratoire, des biopsies tissulaires et des examens d'imagerie. Le traitement vise à gérer les symptômes sous-jacents et à ralentir la progression de la maladie.
La protéine amyloïde A sérique (SAA) est une protéine de phase aiguë, ce qui signifie qu'elle est produite en réponse à une inflammation dans le corps. Elle est synthétisée principalement par le foie et fait partie de la famille des apolipoprotéines associées aux lipoprotéines de haute densité (HDL).
La protéine SAA joue un rôle important dans le système immunitaire, en particulier dans la réponse inflammatoire. Lors d'une infection ou d'une inflammation, les niveaux de SAA augmentent rapidement dans le sang pour aider à combattre l'agent pathogène ou à réparer les tissus endommagés. Cependant, des niveaux élevés et persistants de SAA peuvent contribuer au développement de maladies inflammatoires chroniques et d'autres affections, telles que l'athérosclérose et la polyarthrite rhumatoïde.
Dans certaines conditions, comme la maladie de l'alimentation amyloïde, la protéine SAA peut se déposer dans les tissus sous forme d'amyloïdes, des agrégats fibrillaires insolubles qui peuvent perturber leur fonction et entraîner des dommages organiques. Ces dépôts amyloïdes sont souvent associés à une inflammation chronique et à la production accrue de protéines SAA.
La préalbumine, également connue sous le nom de transthyrétine, est une protéine présente dans le sang et la cavité abdominale. Elle joue un rôle important dans le transport des hormones thyroïdiennes et de la vitamine A dans l'organisme.
Dans un contexte médical, les taux de préalbumine peuvent être utilisés comme marqueur de la nutrition et de la fonction hépatique d'un patient. Les niveaux de préalbumine sont souvent corrélés avec l'état nutritionnel général d'une personne, car ils ont tendance à diminuer en cas de malnutrition ou de maladies inflammatoires chroniques.
Les taux de préalbumine peuvent être mesurés par une analyse de sang et sont souvent utilisés en combinaison avec d'autres marqueurs pour évaluer l'état nutritionnel et la gravité de certaines maladies, telles que la maladie inflammatoire de l'intestin ou le cancer.
Il est important de noter que des facteurs tels que l'âge, le sexe, l'origine ethnique et les conditions médicales préexistantes peuvent influencer les niveaux de préalbumine d'un individu. Par conséquent, les résultats doivent être interprétés en tenant compte du contexte clinique global du patient.
Les chaînes légères des immunoglobulines sont des protéines structurelles importantes qui composent les anticorps, également connus sous le nom d'immunoglobulines. Il existe deux types de chaînes légères : kappa (κ) et lambda (λ). Chaque anticorps est constitué de deux chaînes lourdes et deux chaînes légères, qui sont reliées entre elles par des ponts disulfure pour former la structure tridimensionnelle de l'anticorps.
Les chaînes légères sont synthétisées sur les ribosomes libres du réticulum endoplasmique et contiennent deux domaines constants (C) et un domaine variable (V). Le domaine variable est responsable de la reconnaissance et de la liaison aux antigènes, tandis que les domaines constants sont responsables de la fonction effectrice de l'anticorps.
Les chaînes légères kappa et lambda sont codées par des gènes différents situés sur des chromosomes différents. Chez un individu sain, il y a une distribution équilibrée des chaînes légères kappa et lambda dans le sang et la moelle osseuse. Cependant, certaines affections médicales peuvent entraîner une production anormale de chaînes légères, telles que les gammapathies monoclonales, qui sont des conditions caractérisées par la production excessive d'une seule chaîne légère. Ces conditions peuvent être bénignes ou malignes et nécessitent une évaluation médicale appropriée.
La Fièvre Méditerranéenne Familiale (FMF) est une maladie héréditaire rare associée à des épisodes récurrents de fièvre, douleurs abdominales, inflammation articulaire et éruptions cutanées. Elle est causée par des mutations dans le gène MEFV qui code pour la protéine pyrin. Cette protéine joue un rôle important dans la régulation de l'inflammation. Les mutations du gène MEFV entraînent une activation excessive et inappropriée du système immunitaire, conduisant aux symptômes caractéristiques de la FMF.
Les épisodes de fièvre peuvent durer de quelques heures à plusieurs jours et sont souvent accompagnés de douleurs abdominales sévères qui peuvent être confondues avec une appendicite aiguë. L'inflammation articulaire peut toucher n'importe quelle articulation, mais elle est le plus souvent observée dans les genoux et les chevilles. Les éruptions cutanées sont généralement des plaques rouges et douloureuses qui apparaissent sur les jambes et les bras.
Le diagnostic de la FMF repose sur les antécédents cliniques, l'examen physique et les résultats des tests de laboratoire. La confirmation du diagnostic nécessite souvent une analyse génétique pour détecter les mutations dans le gène MEFV.
Le traitement de la FMF repose sur l'utilisation d'anti-inflammatoires non stéroïdiens (AINS) pour soulager les symptômes pendant les épisodes aigus, et des médicaments modificateurs de la réponse biologique (MRB), tels que le colchicine, pour prévenir les récidives. Dans les cas graves ou résistants au traitement, d'autres options thérapeutiques peuvent être envisagées, telles que l'interleukine-1 (IL-1) inhibiteurs.
Les neuropathies amyloïdes sont un groupe de troubles nerveux causés par la présence de dépôts anormaux d'une protéine appelée "amyloïde" dans les tissus nerveux. Ces dépôts peuvent endommager et détruire les nerfs, entraînant une variété de symptômes neurologiques.
Les neuropathies amyloïdes peuvent affecter les nerfs sensoriels, moteurs ou autonomes, entraînant des symptômes tels que des picotements, des engourdissements, des douleurs, une faiblesse musculaire, une perte de réflexes, des problèmes d'équilibre et de coordination, des troubles digestifs, cardiaques ou urinaires.
Les neuropathies amyloïdes peuvent être héréditaires ou acquises. Les formes héréditaires sont souvent causées par des mutations dans les gènes qui produisent les protéines amyloïdes, tandis que les formes acquises peuvent être associées à des maladies sous-jacentes telles que le myélome multiple, la maladie de Waldenström ou l'insuffisance rénale chronique.
Le diagnostic de neuropathies amyloïdes repose sur une combinaison d'examens cliniques, électrophysiologiques et de biopsie nerveuse pour confirmer la présence de dépôts amyloïdes dans les tissus nerveux. Le traitement dépend de la cause sous-jacente et peut inclure des médicaments pour soulager les symptômes, des thérapies physiques ou des greffes de cellules souches.
Je suis désolé, mais il n'y a pas de terme médical communément reconnu appelé "Rouge Congo". Il est possible que vous fassiez référence à un type de teinture ou de colorant chimique connu sous le nom de " Rouge de Congo ". Cependant, ce n'est pas non plus un terme directement lié au domaine médical. Le Rouge de Congo est une teinture rouge brillante utilisée en microscopie pour la coloration des tissus et des cellules. Il est utilisé pour mettre en évidence certaines structures, telles que les mastocytes et les fibres élastiques. Mais rappelez-vous qu'il s'agit d'un usage histologique et non médical direct.
Les neuropathies amyloïdes familiales sont un groupe de troubles nerveux héréditaires rares causés par des mutations génétiques spécifiques. Ces mutations entraînent la production d'une protéine anormale qui a tendance à s'agréger et à former des dépôts amyloïdes dans divers tissus du corps, y compris les nerfs périphériques.
Les neuropathies amyloïdes familiales peuvent affecter à la fois le système nerveux périphérique (les nerfs en dehors du cerveau et de la moelle épinière) et le système nerveux autonome (qui contrôle les fonctions automatiques du corps telles que la fréquence cardiaque, la pression artérielle, la digestion et la température corporelle).
Les symptômes de ces neuropathies peuvent inclure une perte de sensation, des picotements ou un engourdissement dans les mains et les pieds, des douleurs nerveuses, une faiblesse musculaire, des problèmes d'équilibre et de coordination, ainsi que des problèmes digestifs, cardiaques et urinaires dus à l'atteinte du système nerveux autonome.
Il existe plusieurs types de neuropathies amyloïdes familiales, chacune étant causée par une mutation génétique différente. Les traitements peuvent inclure des médicaments pour soulager les symptômes, la physiothérapie et, dans certains cas, des thérapies expérimentales telles que la transplantation de cellules souches ou l'administration d'anticorps monoclonaux.
Les cardiomyopathies sont des maladies du muscle cardiaque (myocarde) qui affectent sa structure et sa fonction, entraînant une incapacité à pomper le sang efficacement dans tout l'organisme. Il existe différents types de cardiomyopathies, chacune ayant des causes, des symptômes et des traitements variés. Les principaux types comprennent:
1. Cardiomyopathie dilatée (CMD): Dans ce type, le ventricule gauche ou les deux ventricules du cœur s'affaiblissent et s'élargissent, ce qui entraîne une réduction de la capacité de pompage du cœur. La CMD peut être héréditaire ou causée par des facteurs tels que l'insuffisance cardiaque, l'hypertension artérielle, l'alcoolisme et certaines infections virales.
2. Cardiomyopathie hypertrophique (CMH): Dans la CMH, le muscle cardiaque devient anormalement épais, rigide et difficile à étirer, ce qui rend plus difficile pour le cœur de remplir correctement entre les battements. Cette condition est souvent héréditaire mais peut également être causée par des maladies telles que l'hypertension artérielle et la maladie de Fabry.
3. Cardiomyopathie restrictive (CMR): Dans ce type, le muscle cardiaque devient rigide et incapable de se remplir correctement entre les battements, entraînant une réduction de la capacité de pompage du cœur. La CMR peut être causée par des affections sous-jacentes telles que l'amylose, la sclérodermie et certaines maladies hépatiques.
4. Cardiomyopathie arythmogène droite du ventricule (CAM): Dans ce type, les cellules musculaires du ventricule droit sont remplacées par des tissus cicatriciels et graisseux, entraînant une altération de la capacité de pompage du cœur. La CAM est souvent héréditaire et peut être associée à des arythmies cardiaques graves.
Les symptômes de ces différents types de cardiomyopathie peuvent varier considérablement, allant de presque aucun symptôme à une insuffisance cardiaque grave ou même à la mort subite. Le traitement dépend du type et de la gravité de la maladie et peut inclure des médicaments, des dispositifs implantables tels que des défibrillateurs cardioverteurs implantables (ICD) ou des stimulateurs cardiaques, ainsi qu'une intervention chirurgicale potentielle. Dans certains cas, une transplantation cardiaque peut être recommandée.
La bêta-2-microglobuline est une protéine qui se trouve à la surface de certaines cellules, en particulier les globules blancs appelés lymphocytes T et B. Elle fait partie du complexe majeur d'histocompatibilité de classe I, qui joue un rôle important dans le système immunitaire en présentant des antigènes aux cellules T pour qu'elles puissent les reconnaître et y répondre.
La bêta-2-microglobuline peut également être trouvée dans le sang et l'urine à des niveaux très faibles, où elle est utilisée comme un marqueur de la fonction rénale et de certaines maladies, telles que les maladies inflammatoires et les cancers du sang. Des niveaux élevés de bêta-2-microglobuline dans le sang peuvent indiquer une mauvaise fonction rénale ou une production accrue de cellules anormales, comme c'est le cas dans la maladie de Hodgkin, le myélome multiple et d'autres troubles hématologiques.
Dans le diagnostic et le suivi des maladies malignes hématologiques, les taux sériques de bêta-2-microglobuline sont couramment utilisés pour évaluer la charge tumorale et l'efficacité du traitement. Des niveaux élevés de bêta-2-microglobuline peuvent également être associés à une mauvaise pronostique dans ces maladies.
Le composant amyloïde P sérique, également connu sous le nom de sAP, est une forme soluble du peptide bêta-amyloïde (Aβ) qui circule dans le sang. Les peptides bêta-amyloïdes sont des fragments de protéines précurseurs amyloïdes (APP) qui sont normalement présents dans le cerveau et jouent un rôle important dans la structure et la fonction des neurones.
Cependant, lorsque les peptides bêta-amyloïdes s'agglutinent et forment des plaques amyloïdes, ils peuvent être déposés anormalement dans le cerveau, entraînant une accumulation toxique qui peut endommager et tuer les neurones. Cette accumulation est un signe distinctif de la maladie d'Alzheimer et d'autres troubles neurodégénératifs.
Le composant amyloïde P sérique est mesuré dans le sang comme un biomarqueur possible pour détecter et surveiller la maladie d'Alzheimer et d'autres troubles neurodégénératifs. Des niveaux élevés de sAP peuvent indiquer une accumulation accrue de plaques amyloïdes dans le cerveau, ce qui peut aider au diagnostic précoce et à la surveillance de la maladie. Cependant, des recherches supplémentaires sont nécessaires pour déterminer si les niveaux de sAP peuvent être utilisés comme un biomarqueur fiable et sensible pour le diagnostic et la surveillance de la maladie d'Alzheimer et d'autres troubles neurodégénératifs.
Le syndrome néphrotique est un trouble rénal caractérisé par une combinaison de symptômes et de signes résultant d'une altération de la barrière de filtration glomérulaire. Il se traduit principalement par une protéinurie sévère (perte excessive de protéines dans les urines, généralement supérieure à 3,5 g par jour), une hypoalbuminémie sévère (diminution du taux d'albumine dans le sang) et une hyperlipidémie (augmentation des lipides sanguins).
Ce syndrome peut entraîner un œdème, qui est une accumulation de liquide dans les tissus corporels, en particulier au niveau des membres inférieurs et du visage. Il peut également conduire à une susceptibilité accrue aux infections, en raison de la baisse des niveaux d'immunoglobulines dans le sang.
Le syndrome néphrotique peut être primitif (idiopathique), ce qui signifie qu'il est la conséquence directe d'une maladie rénale sous-jacente, ou secondaire, lorsqu'il est associé à une autre pathologie systémique, comme le diabète sucré, l'amylose, les vascularites, ou certaines infections.
Les causes primitives du syndrome néphrotique sont souvent liées à des lésions glomérulaires spécifiques, telles que la néphrose lipoïde (associée à une atteinte des podocytes) ou la glomérulosclérose segmentaire et focale. Le diagnostic repose sur l'analyse de l'urine, du sang et parfois sur une biopsie rénale pour identifier la cause sous-jacente et adapter le traitement.
Les paraprotéinémies sont des conditions médicales dans lesquelles il y a une production excessive d'une protéine anormale appelée chaîne légère d'immunoglobuline ou paraprotéine. Ces protéines sont produites par certaines cellules du système immunitaire, appelées plasmocytes, qui se trouvent principalement dans la moelle osseuse.
Les paraprotéinémies peuvent être associées à une variété de maladies, y compris les troubles sanguins et les cancers, tels que le myélome multiple, le macroglobulinémie de Waldenström, et certains types de lymphomes. Dans ces conditions, la production excessive de paraprotéines peut entraîner une gamme de symptômes, y compris la fatigue, des saignements, des infections fréquentes, des douleurs osseuses, et des dommages aux organes internes.
Il est important de noter que toutes les personnes atteintes de paraprotéinémies ne présenteront pas nécessairement de symptômes ou de maladies sous-jacentes associées. Dans certains cas, la découverte d'une paraprotéinémie peut être fortuite et ne nécessiter aucun traitement spécifique. Cependant, il est important de surveiller régulièrement les personnes atteintes de paraprotéinémies pour détecter tout signe de maladie sous-jacente ou de complications associées à la production excessive de paraprotéines.
La "maladie de la trachée" est un terme général qui se réfère à diverses affections, infections, ou conditions qui affectent la trachée, qui est la voie respiratoire principale qui transporte l'air inspiré vers les poumons. Voici quelques exemples de maladies de la trachée :
1. La sténose trachéale : un rétrécissement anormal de la lumière de la trachée, souvent causé par une cicatrice ou une tumeur.
2. La trachéite : une inflammation de la muqueuse trachéale, généralement causée par une infection virale ou bactérienne.
3. Le traumatisme de la trachée : des blessures à la trachée peuvent être causées par un traumatisme direct, comme un coup violent sur le cou, ou par un traumatisme indirect, comme une intubation difficile.
4. Les tumeurs de la trachée : les tumeurs bénignes et malignes peuvent se développer dans la trachée et entraîner des symptômes respiratoires.
5. La trichobézoaire trachéale : une accumulation anormale de cheveux ou de poils dans la trachée, souvent observée chez les personnes atteintes du syndrome de Rapunzel.
6. Les malformations congénitales de la trachée : des anomalies structurelles de la trachée peuvent être présentes à la naissance et entraîner des problèmes respiratoires.
Le traitement d'une maladie de la trachée dépend de la cause sous-jacente et peut inclure des médicaments, une intervention chirurgicale, ou une combinaison des deux.
Les maladies rénales sont des affections ou des désordres qui affectent les reins et perturbent leur fonction normale d'élimination des déchets et des liquides du corps. Elles peuvent être causées par une variété de facteurs, y compris des infections, des malformations congénitales, des maladies auto-immunes, des traumatismes, des tumeurs et des affections systémiques telles que le diabète et l'hypertension artérielle.
Les maladies rénales peuvent se manifester de diverses manières, selon leur cause sous-jacente et leur gravité. Les symptômes courants comprennent une urination fréquente ou douloureuse, une fatigue extrême, des douleurs lombaires, un gonflement des jambes et des chevilles, une hypertension artérielle et une mauvaise haleine.
Les maladies rénales peuvent entraîner des complications graves, telles que l'insuffisance rénale, qui peut nécessiter une dialyse ou une greffe de rein. Il est donc important de diagnostiquer et de traiter les maladies rénales dès que possible pour prévenir des dommages irréversibles aux reins.
Le diagnostic des maladies rénales implique généralement des tests d'imagerie, tels qu'une échographie ou une tomographie computérisée, ainsi que des analyses d'urine et de sang pour évaluer la fonction rénale. Le traitement dépend de la cause sous-jacente de la maladie rénale et peut inclure des médicaments, des changements de mode de vie ou une intervention chirurgicale.
La macroglossie est un terme médical qui se réfère à une langue anormalement grande ou hypertrophiée. Cette condition peut être congénitale, c'est-à-dire présente à la naissance, ou acquise plus tard dans la vie en raison de diverses causes.
La macroglossie congénitale est souvent associée à certaines maladies génétiques telles que le syndrome de Down, le syndrome de Beckwith-Wiedemann et d'autres troubles de croissance et développement.
D'un autre côté, la macroglossie acquise peut être due à des causes variées telles que des affections inflammatoires ou infectieuses (comme l'amylose, la maladie de Riedel, la diphtérie), des tumeurs bénignes ou malignes de la langue, une réaction médicamenteuse, un œdème lymphatique chronique, ou encore un état de manque d'hormones thyroïdiennes (hypothyroïdie).
La macroglossie peut causer divers problèmes, y compris des difficultés à manger, à parler et à avaler; une respiration difficile, surtout pendant le sommeil (ce qui peut entraîner une apnée obstructive du sommeil); des fissures ou des plaies douloureuses sur la surface de la langue; et dans certains cas, une apparence faciale anormale. Le traitement dépend de la cause sous-jacente et peut inclure des médicaments, une intervention chirurgicale, une thérapie de radiation ou une combinaison de ces options.
Les protéines amyloïdogènes sont un type de protéines qui ont la capacité de se plier et de s'agréger de manière anormale, formant des structures bêta-plissées riches en feuillets β qui sont caractéristiques des dépôts amyloïdes. Ces dépôts peuvent s'accumuler dans divers tissus et organes du corps, entraînant une variété de maladies connues sous le nom de maladies amyloïdoses.
Les protéines amyloïdogènes comprennent plusieurs types différents, dont la protéine bêta-amyloïde dans la maladie d'Alzheimer, la protéine transthyrétine dans l'amylose cardiaque et la protéine lysozyme dans l'amylose rénale. Ces protéines sont normalement solubles et remplissent des fonctions importantes dans l'organisme, mais lorsqu'elles sont mal pliées ou dénaturées, elles peuvent s'agréger et former des dépôts amyloïdes qui perturbent la fonction normale des tissus et des organes.
Le processus d'agrégation des protéines amyloïdogènes est complexe et peut être influencé par une variété de facteurs, y compris les mutations génétiques, l'âge, le stress oxydatif et d'autres facteurs environnementaux. La compréhension du processus d'agrégation des protéines amyloïdogènes est un domaine de recherche actif dans le développement de nouveaux traitements pour les maladies amyloïdoses.
Melphalan est un médicament de chimiothérapie utilisé dans le traitement de certains types de cancer, y compris le myélome multiple et le cancer des ovaires. Il appartient à une classe de médicaments appelés agents alkylants qui travaillent en interférant avec l'ADN du cancer, ce qui entraîne des dommages à l'ADN et empêche la croissance et la multiplication des cellules cancéreuses.
Melphalan est disponible sous forme de comprimés ou d'une solution injectable et est généralement administré dans un cadre hospitalier ou clinique. Les effets secondaires courants du melphalan peuvent inclure la nausée, la diarrhée, la fatigue, la perte de cheveux et une diminution du nombre de cellules sanguines. Des effets secondaires plus graves peuvent inclure des lésions pulmonaires, des dommages au foie et un risque accru d'infections.
Comme avec tous les médicaments de chimiothérapie, le melphalan peut également affecter les cellules sanguines normales, ce qui entraîne une diminution du nombre de globules blancs, de plaquettes et de globules rouges. Cela peut augmenter le risque d'infections, de saignements et d'anémie. Les patients recevant du melphalan doivent faire l'objet d'une surveillance étroite pour détecter les signes d'effets secondaires et de complications.
La protéine Bence Jones est un type spécifique de chaîne légère d'immunoglobuline monoclonale trouvée dans l'urine ou parfois dans le sérum sanguin. Elle est nommée d'après le médecin britannique Henry Bence Jones qui l'a décrite pour la première fois en 1847. Ces chaînes légères sont produites en excès par certaines cellules tumorales, principalement dans les myélomes multiples et les maladies des chaînes légères (une forme de gammapathie monoclonale).
Les protéines Bence Jones peuvent être détectées par des tests d'urine ou de sang. Lorsqu'elles sont chauffées puis refroidies, elles forment des précipités, ce qui est utilisé comme méthode de détection en laboratoire. L'accumulation de ces protéines peut entraîner des dommages aux reins et d'autres complications liées à la maladie sous-jacente.
Une biopsie est un procédé médical dans lequel un échantillon de tissu corporel est prélevé et examiné de près, généralement au microscope, pour déterminer la présence ou l'absence d'une maladie ou d'une condition spécifique. Ce processus aide les médecins à poser un diagnostic précis et à planifier le traitement approprié.
Il existe plusieurs types de biopsies, en fonction de la partie du corps concernée et de la méthode utilisée pour prélever l'échantillon :
1. Biopsie par incision/excision : Dans ce type de biopsie, une incision est pratiquée dans la peau pour accéder au tissu cible. L'échantillon peut être retiré en entier (biopsie excisionnelle) ou seulement une partie du tissu peut être prélevée (biopsie incisionnelle). Ce type de biopsie est couramment utilisé pour les grains de beauté suspects, les lésions cutanées et les nodules.
2. Biopsie par aspiration : Cette procédure implique l'utilisation d'une aiguille fine pour prélever un échantillon de tissu ou de liquide dans une zone spécifique du corps. La biopsie à l'aiguille fine est souvent utilisée pour les ganglions lymphatiques enflés, les glandes thyroïdiennes et d'autres organes superficiels.
3. Biopsie au trocart : Dans ce type de biopsie, un trocart (une aiguille creuse) est inséré dans le corps pour prélever un échantillon de tissu. Ce type de biopsie est souvent utilisé pour les organes profonds comme le foie, les poumons et les os.
4. Biopsie chirurgicale : Lors d'une biopsie chirurgicale, une incision est pratiquée dans la peau pour accéder à l'organe ou au tissu cible. Une partie de l'organe ou du tissu est ensuite retirée pour analyse. Ce type de biopsie est souvent utilisé pour les tumeurs malignes et les lésions suspectes dans des organes comme le sein, le poumon et le foie.
5. Biopsie liquide : Cette procédure consiste à analyser un échantillon de sang ou d'autres fluides corporels pour détecter la présence de cellules tumorales ou d'ADN circulant provenant d'une tumeur cancéreuse. La biopsie liquide est une méthode non invasive qui peut être utilisée pour le suivi du traitement et la détection précoce des récidives.
Les résultats de la biopsie sont généralement examinés par un pathologiste, qui fournit un rapport décrivant les caractéristiques du tissu ou des cellules prélevés. Ce rapport peut aider à déterminer le diagnostic et le traitement appropriés pour une maladie spécifique.
'Acinonyx' est un genre de félidés qui ne compte qu'une seule espèce actuelle : le guépard (*Acinonyx jubatus*). Contrairement aux autres grands félins, les guépards sont caractérisés par leur incapacité à rétracter leurs griffes et leur adaptation à la course à grande vitesse.
Les guépards sont des animaux fascinants avec des adaptations uniques pour la chasse au gibier rapide. Leur corps mince, musclé et flexible, ainsi que leurs longues pattes postérieures, leur permettent d'atteindre des vitesses de pointe allant jusqu'à 105 kilomètres à l'heure (65 miles par heure).
Cependant, les guépards sont également considérés comme vulnérables sur la liste rouge de l'UICN en raison de la perte d'habitat et de la diminution des populations de proies. Leur population est fragmentée dans toute leur aire de répartition, qui s'étend sur une grande partie de l'Afrique subsaharienne et une petite partie de l'Asie centrale.
Il convient de noter que le terme 'Acinonyx' fait référence au genre taxonomique du guépard et ne doit pas être confondu avec un terme médical spécifique.
Les chaînes légères lambda des immunoglobulines sont des protéines structurelles importantes qui composent les anticorps, également connus sous le nom d'immunoglobulines. Les anticorps sont des molécules clés du système immunitaire qui aident à identifier et à neutraliser les agents pathogènes étrangers tels que les bactéries et les virus.
Chaque anticorps est composé de deux chaînes lourdes et deux chaînes légères, qui sont reliées entre elles par des ponts disulfures pour former une structure en Y. Les chaînes légères peuvent être divisées en deux types différents : kappa et lambda. Environ 60% des anticorps humains contiennent des chaînes légères lambda, tandis que les 40% restants contiennent des chaînes légères kappa.
Les chaînes légères lambda sont codées par un gène situé sur le chromosome 22. Les mutations ou les réarrangements anormaux de ce gène peuvent entraîner la production d'un nombre anormal de chaînes légères lambda, ce qui peut contribuer au développement de certaines maladies du sang telles que le myélome multiple et le trouble du gène WM (Waldenström macroglobulinémie).
En résumé, les chaînes légères lambda des immunoglobulines sont des protéines structurelles importantes qui composent les anticorps et aident à identifier et à neutraliser les agents pathogènes étrangers. Les anomalies dans la production de ces chaînes peuvent contribuer au développement de certaines maladies du sang.
La gelsoline est une protéine actine-liant qui joue un rôle crucial dans la restructuration des filaments d'actine, ce qui en fait un élément essentiel du maintien de la structure cellulaire et de la mobilité cellulaire. Elle a la capacité de séparer les filaments d'actine en courts fragments, un processus connu sous le nom de coupage, et peut également favoriser l'assemblage de nouveaux filaments. La gelsoline est également capable de se lier à des membranes riches en phosphatidylinositol 4,5-bisphosphate (PIP2), ce qui permet une localisation et une activation spécifiques au site dans la cellule. Des déséquilibres ou des mutations dans l'expression de la gelsoline ont été associés à un certain nombre de conditions pathologiques, telles que les maladies neurodégénératives, les maladies cardiovasculaires et certains types de cancer.
Le déficit en facteur X est un trouble hémorragique rare causé par une mutation génétique qui entraîne une diminution de l'activité du facteur X, une protéine essentielle dans la coagulation sanguine. Ce facteur joue un rôle clé dans la conversion de la prothrombine en thrombine, une étape cruciale dans la formation d'un caillot sanguin pour arrêter le saignement.
Les personnes atteintes de ce déficit peuvent présenter des saignements spontanés ou prolongés après une blessure, une intervention chirurgicale ou l'accouchement. Les symptômes varient en fonction du niveau de facteur X fonctionnel dans le sang. Les cas sévères peuvent provoquer des hémorragies graves dès la naissance, tandis que les cas plus légers peuvent ne présenter des saignements qu'à l'âge adulte, souvent déclenchés par un traumatisme ou une intervention médicale.
Le diagnostic repose généralement sur des tests de coagulation sanguine spécifiques qui mesurent l'activité du facteur X. Le traitement consiste souvent en des perfusions régulières de concentré de facteur X pour prévenir et contrôler les saignements. Une surveillance attentive par un hématologue est nécessaire pour assurer une thérapie adéquate et éviter les complications, telles que l'accumulation excessive de caillots sanguins (thrombose).
Les maladies génétiques de la peau sont des affections cutanées héréditaires causées par des mutations dans les gènes. Ces mutations peuvent être transmises des parents à l'enfant ou peuvent survenir spontanément pendant le développement de l'embryon. Les maladies génétiques de la peau affectent la structure, la fonction et l'apparence de la peau, entraînant souvent des symptômes cutanés visibles ainsi que d'autres complications systémiques.
Les exemples courants de maladies génétiques de la peau comprennent :
1. La neurofibromatose : une affection caractérisée par la croissance de tumeurs bénignes le long des nerfs, entraînant des lésions cutanées et des complications neurologiques.
2. Le xeroderma pigmentosum : un trouble rare qui rend la peau extrêmement sensible aux rayons UV, entraînant un risque élevé de cancer de la peau et d'autres dommages cutanés.
3. La maladie de Darier : une maladie héréditaire de la kératinisation qui provoque des plaques squameuses et des démangeaisons sur la peau, en particulier dans les zones humides et chaudes du corps.
4. L'ichtyose : un groupe de troubles génétiques qui affectent la différenciation des kératinocytes, entraînant une accumulation de squames et une peau sèche et écailleuse.
5. Le syndrome d'Ehlers-Danlos : un trouble du tissu conjonctif caractérisé par une peau fine, élastique et fragile, ainsi que des articulations hypermobiles et des complications vasculaires.
6. La maladie de Hurler : une maladie héréditaire rare du métabolisme des mucopolysaccharides qui affecte la peau, les os, les yeux, le cœur et d'autres organes.
7. Le syndrome de Marfan : un trouble génétique du tissu conjonctif qui affecte la croissance et le développement des os, des yeux, du cœur et des vaisseaux sanguins.
8. La neurofibromatose : un groupe de troubles héréditaires caractérisés par la formation de tumeurs bénignes sur les nerfs et la peau.
9. Le syndrome de Peutz-Jeghers : une maladie génétique rare qui se caractérise par des taches pigmentées sur la peau et les muqueuses, ainsi que des polypes intestinaux bénins.
10. La xeroderma pigmentosum : un trouble héréditaire rare de la réparation de l'ADN qui entraîne une sensibilité extrême aux rayons UV et un risque élevé de cancer de la peau.
Un résultat fatal en médecine se réfère à un décès ou au fait de causer la mort. C'est un terme utilisé pour décrire un résultat particulièrement grave d'une maladie, d'un traumatisme ou d'une procédure médicale. Un résultat fatal peut être attendu dans certaines situations, comme dans le cas de maladies avancées et terminaux, ou peut être imprévu et survenir même avec un traitement approprié. Dans les deux cas, il s'agit d'un événement tragique qui a des implications importantes pour les patients, leurs familles et les professionnels de la santé qui les prennent en charge.
Le myélome multiple, également connu sous le nom de plasmocytome malin multiple ou encore cancer des plasmocytes, est un type de cancer qui se forme dans les cellules plasmatiques du système immunitaire. Ces cellules sont normalement responsables de la production d'anticorps pour aider à combattre les infections.
Dans le myélome multiple, les cellules cancéreuses s'accumulent dans la moelle osseuse, où elles peuvent affaiblir et fragiliser les os, entraînant des douleurs osseuses, des fractures et une augmentation du risque de développer des infections. Les cellules cancéreuses peuvent également s'accumuler dans d'autres parties du corps, telles que le rein, ce qui peut entraîner une insuffisance rénale.
Les symptômes courants du myélome multiple comprennent la fatigue, des douleurs osseuses, des infections fréquentes, une anémie, une perte de poids et une augmentation de la calcémie (taux élevé de calcium dans le sang). Le diagnostic est généralement posé à l'aide d'une biopsie de la moelle osseuse et d'analyses de sang pour déterminer la présence de protéines anormales produites par les cellules cancéreuses.
Le traitement du myélome multiple peut inclure une combinaison de chimiothérapie, de radiothérapie, de thérapies ciblées, d'immunothérapie et de greffe de moelle osseuse. Le pronostic dépend du stade de la maladie au moment du diagnostic, de l'âge du patient et de sa réponse au traitement.
La troponine est un complexe de protéines trouvé dans les muscles striés, y compris le muscle cardiaque. Il joue un rôle crucial dans la régulation de la contraction musculaire. Dans le contexte médical, les taux de troponines sont fréquemment mesurés comme marqueurs pour évaluer les dommages au muscle cardiaque.
Il existe trois types de troponines: troponine T, troponine I et troponine C. Les troponines T et I sont spécifiques au muscle cardiaque (c'est-à-dire myocarde), tandis que la troponine C est présente dans tous les types de muscles striés. Lorsqu'il y a une nécrose (mort) des cellules musculaires cardiaques, comme c'est le cas dans une crise cardiaque (infarctus du myocarde), ces protéines sont libérées dans la circulation sanguine.
Des niveaux élevés de troponines T ou I dans le sang indiquent donc généralement des dommages au muscle cardiaque. Ces marqueurs sont très sensibles et spécifiques, ce qui signifie qu'ils peuvent détecter même de petites quantités de lésions myocardiques et aider à distinguer les dommages cardiaques des dommages à d'autres types de muscles.
En résumé, la troponine est une protéine musculaire cardiaque utilisée comme marqueur pour diagnostiquer et évaluer l'étendue des lésions myocardiques dans diverses conditions telles que les crises cardiaques.
La troponine C est un composant des complexes de protéines régulatrices de la contraction musculaire trouvés dans les muscles squelettiques et cardiaques. Elle joue un rôle crucial dans la régulation calcium-dépendante de la contraction et relaxation musculaires. Dans le diagnostic médical, les niveaux de troponine C dans le sang peuvent être mesurés pour évaluer les dommages myocardiques, en particulier dans le contexte d'un infarctus du myocarde (crise cardiaque). Les tests de troponine sont souvent utilisés en combinaison avec d'autres examens pour déterminer l'étendue des dommages au muscle cardiaque et guider la prise en charge thérapeutique. Cependant, il est important de noter que la troponine C n'est pas spécifique aux dommages myocardiques, car elle peut également être libérée dans le sang en cas de lésions musculaires squelettiques sévères.
La troponine T est une protéine régulatrice de la contraction musculaire trouvée dans les cardiomyocytes, ou cellules musculaires cardiaques. Elle fait partie du complexe troponine, avec la troponine I et la troponine C. Lorsque le muscle cardiaque est endommagé, comme dans une crise cardiaque (infarctus du myocarde), les troponines T, I et C sont libérées dans le sang.
En particulier, la troponine T est considérée comme un marqueur très sensible et spécifique des dommages au muscle cardiaque. Les niveaux de troponine T dans le sang peuvent être mesurés pour diagnostiquer et évaluer l'étendue des dommages causés par une crise cardiaque. Des niveaux élevés de troponine T sont également associés à d'autres affections cardiovasculaires, telles que l'insuffisance cardiaque congestive et les arythmies cardiaques.
Il est important de noter que des niveaux élevés de troponine T ne signifient pas nécessairement qu'une personne a eu une crise cardiaque, mais plutôt qu'il y a eu un dommage au muscle cardiaque. D'autres facteurs, tels que l'exercice intense ou les maladies musculaires squelettiques, peuvent également entraîner une augmentation des niveaux de troponine T dans le sang.
Je suis désolé, "Encyclopedias as Topic" n'est pas une définition médicale. Il s'agit plutôt d'une catégorie de sujets dans la classification MeSH (Medical Subject Headings) utilisée pour indexer les articles de bibliographie en médecine et en sciences de la santé. Cette catégorie comprend des encyclopédies médicales générales ou spécialisées, des dictionnaires médicaux, des manuels médicaux et d'autres ressources similaires. Cependant, il ne s'agit pas d'une définition médicale à proprement parler.
Les myosines sont une famille de protéines motrices qui jouent un rôle crucial dans le mouvement et la contraction des muscles squelettiques, lisses et cardiaques. Elles sont également impliquées dans d'autres processus cellulaires tels que la division cellulaire, le transport vésiculaire et la morphogenèse des organites intracellulaires.
Les myosines se composent d'une tête globulaire qui se lie à l'actine, une protéine filamenteuse, et d'une queue allongée qui se lie aux autres structures cellulaires. La tête contient une région catalytique qui hydrolyse l'ATP pour produire de l'énergie, ce qui permet à la myosine de se déplacer le long des filaments d'actine.
Il existe plusieurs types de myosines, chacune ayant des fonctions et des structures spécifiques. Par exemple, la myosine II est responsable de la contraction musculaire, tandis que la myosine V est impliquée dans le transport vésiculaire. Les mutations dans les gènes codant pour les myosines peuvent entraîner des maladies génétiques telles que la cardiomyopathie hypertrophique et la dystrophie musculaire congénitale.
La troponine I est une protéine cardiaque présente dans le muscle cardiaque strié. Elle est libérée dans le sang lorsqu'il y a une nécrose (mort) des cellules musculaires cardiaques, comme c'est le cas lors d'un infarctus du myocarde (crise cardiaque). Les taux sériques de troponine I commencent à augmenter dans les 3 à 6 heures suivant l'infarctus, atteignent un pic après environ 12 à 24 heures et peuvent rester élevés jusqu'à une semaine.
La mesure des taux de troponine I est utilisée en clinique pour diagnostiquer et suivre les lésions myocardiques aiguës. Une élévation des niveaux de troponine I au-dessus des valeurs normales est considérée comme un marqueur sensible et spécifique d'une lésion myocardique aiguë, quelle que soit son étiologie (cause). Cela inclut non seulement les infarctus du myocarde, mais aussi d'autres conditions telles que la myocardite, l'embolie pulmonaire massive et l'insuffisance cardiaque congestive décompensée.
Il est important de noter que des facteurs tels que l'exercice physique intense, les troubles du rythme cardiaque ou la chirurgie cardiaque peuvent également entraîner une augmentation temporaire des taux de troponine I sans qu'il y ait de dommages permanents au muscle cardiaque. Par conséquent, l'interprétation des résultats doit toujours être effectuée en tenant compte du contexte clinique et d'autres tests diagnostiques.
Les muscles sont des organes contractiles qui forment une grande partie du tissu corporel. Ils sont responsables de la mobilité volontaire et involontaire dans le corps humain. Les muscles se contractent pour permettre le mouvement des os, aider à maintenir la posture et contribuer à diverses fonctions corporelles telles que la respiration, la digestion et la circulation sanguine.
Il existe trois types principaux de muscles dans le corps humain :
1. Les muscles squelettiques ou striés : Ils sont attachés aux os par des tendons et leur contraction permet les mouvements volontaires du corps. Ces muscles ont une apparence striée sous un microscope, d'où leur nom.
2. Les muscles lisses : Ce sont des muscles trouvés dans les parois des vaisseaux sanguins, des bronches, de l'utérus et du tube digestif. Ils fonctionnent involontairement, contrôlés par le système nerveux autonome, et participent à des fonctions telles que la circulation, la respiration et la digestion.
3. Le muscle cardiaque : C'est un type spécial de muscle strié qui forme la majeure partie du cœur. Il fonctionne automatiquement sans aucun contrôle volontaire, pompant le sang dans tout le corps.
La capacité des muscles à se contracter et à se détendre provient de leurs propriétés physiques uniques et de la présence de protéines spécialisées telles que l'actine et la myosine, qui glissent les unes contre les autres lorsque le muscle se contracte.
 Troponine
Troponine Besoins d'une mutuelle canine - Santé du chien
Besoins d'une mutuelle canine - Santé du chien HFpEF Flashcards by Nicolas Langevin | Brainscape
HFpEF Flashcards by Nicolas Langevin | Brainscape Fiasp® ultra-fast-acting, PumpCart® (insuline asparte) | Creapharma
Fiasp® ultra-fast-acting, PumpCart® (insuline asparte) | Creapharma AtlasRLeye - Hyalopathie Astéroïde
AtlasRLeye - Hyalopathie Astéroïde Difficulté à respirer (dyspnée): les causes possibles (maladies, traitements...)
Difficulté à respirer (dyspnée): les causes possibles (maladies, traitements...) 23andMe Avis (2023) : La parole des experts
23andMe Avis (2023) : La parole des experts Centre d'imagerie Ostéo-Articulaire | Clinique du sport de Bordeaux
Centre d'imagerie Ostéo-Articulaire | Clinique du sport de Bordeaux Maladie à prions associée à des diarrhées et à une neuropathie du système nerveux autonome - Troubles du cerveau, de la moelle...
Maladie à prions associée à des diarrhées et à une neuropathie du système nerveux autonome - Troubles du cerveau, de la moelle... Français
Français Ressources pour les patient(e)s | CHUM
Ressources pour les patient(e)s | CHUM geneva user, auteur/autrice sur Fondation GEcor
geneva user, auteur/autrice sur Fondation GEcor Mon chat a la patte gonflée - Causes et que faire - GUIDE COMPLET
Mon chat a la patte gonflée - Causes et que faire - GUIDE COMPLET pododermatite plasmocytaire chez le chat - Clinique Alliance
pododermatite plasmocytaire chez le chat - Clinique Alliance APIDRA 100 Unités/ml, solution injectable en cartouche
APIDRA 100 Unités/ml, solution injectable en cartouche Chronique chez le Et. Le - VETINPARIS - VETINPARIS
Chronique chez le Et. Le - VETINPARIS - VETINPARIS Generique Hoodia Moins Cher Deschampsnec - Courses
Generique Hoodia Moins Cher Deschampsnec - Courses Santé | CLINIQUE VÉTÉRINAIRE ACKTARIA
Santé | CLINIQUE VÉTÉRINAIRE ACKTARIA Alzheimer : les anticorps anti-amyloïdes ont-ils un futur ?
Alzheimer : les anticorps anti-amyloïdes ont-ils un futur ? Anavar prohormone, dianabol cycle for beginners - Ниндзя
Anavar prohormone, dianabol cycle for beginners - Ниндзя Veilles - Bibliothèque du CHUM
Veilles - Bibliothèque du CHUM Outils d'Autoguérison
Outils d'Autoguérison buchido | Shar Pei Club de France
buchido | Shar Pei Club de France Luxation du cristallin chez le shar-pei POAG-PLL
Luxation du cristallin chez le shar-pei POAG-PLL