Ataxie
Ataxie Cérébelleuse
Ataxie De Friedreich
Ataxies Spinocérébelleuses
Démarche Ataxique
Ataxie Télangiectasie
Ataxia Telangiectasia Mutated Proteins
Maladie De Machado-Joseph
Expansion Triplets Répétés
Répétition Trinucléotide
Cervelet
Protéines Gène Suppresseur Tumeur
Protéines Cycle Cellulaire
Généalogie
Protein-Serine-Threonine Kinases
Maladies Du Cervelet
Mutation
Troubles De La Motilité Oculaire
Protéines Tissu Nerveux
Syndrome Du Chromosome X Fragile
Apraxies
Age of Onset
Dysarthrie
Protéines Fixant Adn
Protéine Du Syndrome X Fragile
Lésions De L'Adn
Dyssynergie Cérébelleuse Myoclonique
Nystagmus Pathologique
Expansion De Séquences Répétées D'Adn
Canal Potassique Kv1.1
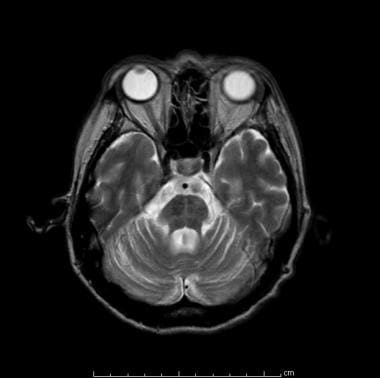
Remnographie
Phénotype
Mutants Neurologiques De Souris
Protéines Nucléaires
Corps D'Inclusion Intranucléaire
Atrophies Olivo-Ponto-Cérébelleuses
Maladies Neurodégénératives Héréditaires
Mutation Faux Sens
Atrophie Multisystématisée
Encéphale
Gliadine
Carence En Vitamine E
Examen Neurologique
Myoclonie
Canaux Calciques Type-P
Maladies Neurodégénératives
Atrophie
Réparation De L'Adn
Maladies Du Système Nerveux
Checkpoint Kinase 2
Rayonnement Ionisant
Dégénérescence Nerveuse
Fasciculation
Syndrome De Miller-Fisher
Hétérozygote
Myoclonic Epilepsies, Progressive
Genetic Linkage
Réflexes Anormaux
Paraplégie Spasmodique Héréditaire
Noyaux Du Cervelet
Epilepsies, Myoclonic
Intellectual Disability
Ophtalmoplégie
Atrophie Optique
Hypoalbuminémie
Phosphotransferases (Alcohol Group Acceptor)
Maladies Expérimentales
Canaux Potassiques Shaw
Allèles
Données Séquence Moléculaire
Cassures Double Brin De L'Adn
L'ataxie est un terme médical utilisé pour décrire une perte de coordination et de contrôle des mouvements musculaires volontaires, ce qui peut entraîner une démarche instable, des tremblements, des mouvements désordonnés et une mauvaise coordination des mains. Cette condition peut affecter les bras, les jambes, les yeux et la parole.
L'ataxie est souvent causée par des dommages au cervelet, qui est la partie du cerveau responsable de la coordination des mouvements, ou aux voies nerveuses qui vont du cervelet au reste du corps. Les causes courantes de l'ataxie comprennent les maladies dégénératives du cerveau telles que la sclérose en plaques, la maladie de Parkinson et la maladie de Charcot-Marie-Tooth, ainsi que les traumatismes crâniens, les tumeurs cérébrales, l'alcoolisme chronique et certains médicaments.
Le traitement de l'ataxie dépend de la cause sous-jacente et peut inclure des médicaments pour contrôler les symptômes, une thérapie physique pour aider à améliorer la coordination et la force musculaire, et des modifications de style de vie pour réduire les facteurs de risque. Dans certains cas, l'ataxie peut être progressive et entraîner une invalidité permanente.
L'ataxie cérébelleuse est un terme utilisé pour décrire un groupe de troubles neurologiques qui affectent le cervelet, une partie du cerveau responsable de la coordination des mouvements volontaires. Les symptômes courants de l'ataxie cérébelleuse comprennent des mouvements corporels maladroits et instables, une démarche chancelante, une perte d'équilibre, une mauvaise coordination des mouvements oculaires, une dysarthrie (parole irrégulière) et des tremblements intentionnels (tremblements qui s'aggravent lorsque vous essayez de faire un mouvement précis).
Les causes de l'ataxie cérébelleuse peuvent varier, mais dans la plupart des cas, elle est causée par une dégénérescence ou une destruction des cellules nerveuses du cervelet. Cette dégénérescence peut être due à une variété de facteurs, y compris des maladies génétiques, des lésions cérébrales, des infections, des tumeurs, des carences nutritionnelles ou l'exposition à des toxines.
Le diagnostic d'ataxie cérébelleuse est généralement posé par un neurologue après avoir évalué les antécédents médicaux du patient, effectué un examen physique et neurologique approfondi, et peut-être commandé des tests d'imagerie ou de laboratoire pour exclure d'autres causes possibles de symptômes.
Le traitement de l'ataxie cérébelleuse dépend de la cause sous-jacente du trouble et peut inclure une combinaison de médicaments, de thérapies de réadaptation, de modifications du mode de vie et d'autres interventions. Dans certains cas, il n'existe pas de traitement curatif pour l'ataxie cérébelleuse, mais les soins de soutien peuvent aider à améliorer la qualité de vie des personnes atteintes de ce trouble.
L'ataxie de Friedreich est une maladie héréditaire rare qui affecte le système nerveux. Elle est causée par une mutation du gène FXN, qui code pour la protéine frataxine. Cette mutation entraîne une accumulation de fer dans les mitochondries des cellules, ce qui endommage progressivement les neurones situés dans la moelle épinière et le cervelet.
Les symptômes de l'ataxie de Friedreich commencent généralement à apparaître entre l'âge de 5 et 15 ans. Les premiers signes comprennent des difficultés à coordonner les mouvements volontaires, une démarche instable et titubante, des tremblements intentionnels (qui s'aggravent lorsque le patient essaie de faire un mouvement), une perte de réflexes profonds, une faiblesse musculaire et une perte de sensation dans les membres inférieurs.
Au fur et à mesure que la maladie progresse, d'autres symptômes peuvent apparaître, tels qu'une paralysie partielle ou complète des jambes, une scoliose (déformation de la colonne vertébrale), une perte auditive et visuelle, un diabète sucré et des problèmes cardiaques.
Actuellement, il n'existe pas de traitement curatif pour l'ataxie de Friedreich. Le traitement est principalement axé sur la gestion des symptômes et peut inclure une thérapie physique pour aider à maintenir la force musculaire et la mobilité, des appareils orthopédiques pour soutenir les membres affaiblis, des médicaments pour traiter les problèmes cardiaques et le diabète sucré, et une assistance respiratoire si nécessaire.
La recherche sur l'ataxie de Friedreich est en cours, avec l'espoir de trouver de nouveaux traitements qui puissent ralentir ou arrêter la progression de la maladie.
Les ataxies spinocérébelleuses (SCAs) représentent un groupe hétérogène de troubles neurologiques progressifs qui affectent le cervelet et la moelle épinière. Le cervelet est une structure située dans le cerveau qui joue un rôle crucial dans la coordination des mouvements volontaires, l'équilibre et les fonctions cognitives. La moelle épinière, quant à elle, est une longue structure nerveuse qui transmet les informations entre le cerveau et le reste du corps.
Les SCAs sont généralement héréditaires et sont causées par des mutations dans divers gènes. Il existe plus de 40 types différents de SCAs, chacun ayant des caractéristiques cliniques et génétiques spécifiques. Les symptômes les plus courants comprennent :
1. Ataxie : Perte de coordination des mouvements volontaires, y compris la marche, l'écriture et la parole.
2. Tremblements : Tremblements intentionnels ou posturaux, qui s'aggravent avec le stress ou l'activité physique.
3. Dysarthrie : Difficulté à articuler les mots en raison d'une faiblesse musculaire ou d'un manque de coordination dans les muscles responsables de la parole.
4. Nystagmus : Mouvements involontaires et oscillatoires des yeux, qui peuvent affecter la vision.
5. Dysphagie : Difficulté à avaler, ce qui peut entraîner un risque accru d'inhalation de nourriture ou de liquides dans les poumons.
6. Faiblesse musculaire : Faiblesse progressive des muscles du tronc et des membres, ce qui peut affecter la capacité à se tenir debout ou à marcher.
7. Perte sensorielle : Diminution de la sensation profonde et de la proprioception (capacité à percevoir la position et les mouvements des articulations), ce qui peut entraîner une démarche instable ou une mauvaise coordination.
8. Problèmes cognitifs : Dans certains cas, des problèmes cognitifs légers peuvent survenir, tels que des difficultés de concentration, de mémoire et d'apprentissage.
Il est important de noter que la progression et la sévérité de ces symptômes varient considérablement d'une personne à l'autre. Certains patients peuvent présenter une ataxie cérébelleuse légère, tandis que d'autres peuvent être gravement touchés par cette maladie.
Ataxia est a general term that refers to a lack of coordination or balance. Ataxic gait, therefore, is a type of abnormal walking pattern characterized by uncoordinated movements and an unsteady, wide-based stance. This can lead to difficulty maintaining balance, stumbling, or falling while walking. Ataxic gait can result from damage to the cerebellum, which is the part of the brain responsible for coordinating muscle movements, or from damage to the sensory nerves that transmit information about body position and movement to the brain.
There are several possible causes of ataxic gait, including:
1. Cerebellar degeneration: This is a condition in which the cerebellum gradually breaks down over time, leading to progressive loss of coordination and balance.
2. Brain injury or tumor: Trauma, infection, stroke, or other conditions that damage the cerebellum can lead to ataxic gait.
3. Multiple sclerosis: This is a chronic autoimmune disease that affects the central nervous system, including the cerebellum.
4. Alcoholism: Long-term alcohol abuse can cause damage to the cerebellum and lead to ataxic gait.
5. Inherited genetic disorders: Certain genetic conditions, such as Friedreich's ataxia or spinocerebellar ataxia, can cause progressive loss of coordination and balance.
Treatment for ataxic gait depends on the underlying cause. Physical therapy, occupational therapy, and assistive devices (such as walkers or canes) may help improve balance and mobility. In some cases, medication or surgery may be necessary to treat the underlying condition.
L'ataxie télangiectasie est une maladie génétique rare et héréditaire qui affecte le système nerveux, le système immunitaire et d'autres systèmes du corps. Elle est causée par des mutations dans un gène spécifique appelé ATM (ataxia telangiectasia mutated).
Les principaux symptômes de l'ataxie télangiectasie comprennent :
1. Ataxie : Il s'agit d'une perte de coordination et de mouvements volontaires, ce qui entraîne une démarche instable et des mouvements corporels maladroits. Cela est dû à une anomalie dans la région cérébelleuse du cerveau.
2. Télangiectasies : Ce sont de petits vaisseaux sanguins dilatés qui se trouvent principalement sur le visage (surtout autour des yeux et des oreilles) et les muqueuses, tels que la bouche et le nez. Ils deviennent généralement apparents vers l'âge de 3 à 6 ans.
3. Problèmes immunitaires : Les personnes atteintes d'ataxie télangiectasie sont sujettes aux infections récurrentes en raison d'un système immunitaire affaibli, en particulier une déficience en anticorps IgG et IgA.
4. Sensibilité accrue au rayonnement : Les personnes atteintes de cette maladie sont plus sensibles aux effets nocifs des radiations, ce qui peut entraîner un risque accru de cancer, en particulier le cancer du sein et du lymphome.
5. Retard de croissance et développement : Les enfants atteints d'ataxie télangiectasie peuvent présenter un retard de croissance et un développement physique et mental.
6. Autres symptômes : D'autres symptômes peuvent inclure des difficultés d'élocution, une mauvaise coordination oculaire, des convulsions et des problèmes endocriniens tels que le diabète insipide.
L'ataxie télangiectasie est une maladie génétique rare, causée par une mutation du gène ATM sur le chromosome 11. Elle se transmet selon un mode autosomique récessif, ce qui signifie que les deux parents doivent être porteurs de la mutation pour que l'enfant développe la maladie. L'espérance de vie moyenne est d'environ 25 à 30 ans, mais cela peut varier considérablement en fonction des complications associées et de la prise en charge médicale.
Ataxia Telangiectasia Mutated (ATM) proteins are crucial in the regulation of the cell cycle and DNA damage response. The ATM gene produces the ATM protein, which is a serine/threonine kinase that plays a central role in detecting DNA double-strand breaks and initiating repair processes.
When DNA damage occurs, ATM proteins become activated and phosphorylate various downstream targets, leading to cell cycle arrest, DNA repair, or apoptosis if the damage is too severe. Mutations in the ATM gene can result in Ataxia Telangiectasia (A-T), a rare autosomal recessive disorder characterized by neurological impairment, immune system dysfunction, increased risk of cancer, and characteristic ocular and cutaneous abnormalities such as telangiectasias.
In summary, ATM proteins are essential for maintaining genomic stability and preventing the development of diseases such as cancer. Defects in these proteins can lead to serious health consequences, including neurological disorders and an increased risk of malignancy.
Les protéines de liaison au fer sont des transporteurs et des stockneurs de fer dans les organismes vivants. Elles jouent un rôle crucial dans la régulation de l'homéostasie du fer, qui est essentielle pour de nombreux processus biologiques tels que la respiration cellulaire, la synthèse de l'ADN et la production d'hormones.
Dans le corps humain, les protéines de liaison au fer comprennent la transferrine, la ferritine et l'hémojuveline. La transferrine est une protéine sérique qui se lie au fer et le transporte dans le sang vers les cellules qui en ont besoin. La ferritine est une protéine de stockage du fer trouvée à la fois dans le cytoplasme des cellules et dans le sérum sanguin, où elle fonctionne comme un tampon pour réguler la concentration de fer libre dans le sang. L'hémojuveline est une protéine de liaison au fer présente dans les membranes plasmiques des cellules qui régulent l'absorption et la libération du fer dans l'intestin grêle.
Un déséquilibre dans les niveaux de protéines de liaison au fer peut entraîner une variété de conditions médicales, telles que l'anémie ferriprive ou l'hémochromatose, qui sont des troubles du métabolisme du fer.
La Maladie de Machado-Joseph, également connue sous le nom de Syndrome spinocérébelleux 3 (SCA3), est une maladie neurodégénérative héréditaire qui affecte le cervelet et la moelle épinière. C'est la forme la plus courante de l'ataxie spinocérébelleuse autosomique dominante dans le monde.
La maladie est causée par une mutation dans le gène ATXN3, qui code pour la protéine ataxine-3. Cette mutation entraîne la production d'une forme anormale de la protéine, qui s'accumule dans les cellules nerveuses du cervelet et de la moelle épinière, provoquant leur mort progressive.
Les symptômes de la maladie de Machado-Joseph comprennent des troubles de l'équilibre et de la coordination, des mouvements oculaires anormaux, une faiblesse musculaire, des tremblements, des difficultés d'élocution et de déglutition, ainsi que des problèmes cognitifs dans les stades avancés de la maladie. Les symptômes apparaissent généralement entre l'âge de 10 et 30 ans, bien que l'apparition plus tardive ne soit pas rare.
Actuellement, il n'existe aucun traitement curatif pour la maladie de Machado-Joseph. Le traitement est principalement axé sur la gestion des symptômes et peut inclure une thérapie physique, une orthophonie, des médicaments pour contrôler les tremblements et d'autres symptômes, ainsi qu'une assistance pour les activités de la vie quotidienne.
Les expansions de triplets répétés (ETR) sont des mutations génétiques dans lesquelles s'observent des séquences d'ADN répétitives. Ces répétitions consistent généralement en une suite de trois nucléotides qui se copient plusieurs fois de manière consécutive. Chez les personnes atteintes d'ETR, ces répétitions sont anormalement longues et peuvent s'allonger encore plus au fil des générations, entraînant une variété de maladies neurodégénératives et autres troubles héréditaires.
Les ETR se produisent le plus souvent dans les régions non codantes de l'ADN, qui ne contiennent pas d'instructions pour la production de protéines. Cependant, lorsque ces répétitions se trouvent dans des gènes spécifiques, elles peuvent perturber la fonction génique et entraîner une maladie.
Les ETR sont associés à un large éventail de troubles héréditaires, notamment :
1. Maladie de Huntington - Une maladie neurodégénérative qui affecte le mouvement, la cognition et le comportement.
2. Ataxies spinocérébelleuses (SCA) - Un groupe de troubles du cervelet qui affectent l'équilibre, la coordination et les mouvements volontaires.
3. Myotonie de Steinert - Une maladie neuromusculaire caractérisée par une raideur musculaire et des crampes.
4. Maladie de la fragile X - Un trouble du développement qui peut causer des retards intellectuels, des problèmes d'élocution et des traits physiques distinctifs.
5. Dystrophie myotonique de type 1 (DM1) - Une maladie neuromusculaire caractérisée par une faiblesse musculaire progressive, une cataracte et des problèmes cardiaques.
Le nombre de répétitions d'une séquence nucléotidique spécifique dans un gène est directement lié à la gravité et à l'âge d'apparition de ces maladies. Plus le nombre de répétitions est élevé, plus les symptômes sont graves et plus ils apparaissent tôt dans la vie. Ce phénomène est connu sous le nom d'expansion de la répétition des trinucléotides.
Une répétition trinucléotide est un type spécifique de mutation génétique dans laquelle une séquence de trois nucléotides (les unités de base de l'ADN) est répétée de manière anormale et excessive dans une région donnée du gène. Normalement, ces répétitions sont présentes en petit nombre dans le génome, mais lorsqu'elles sont multipliées, elles peuvent entraîner des modifications de la structure et de la fonction des protéines codées par les gènes concernés.
Les répétitions trinucléotides sont souvent associées à certaines maladies neurodégénératives héréditaires, telles que la maladie de Huntington, la maladie de Charcot-Marie-Tooth et la sclérose latérale amyotrophique spinale (SLA) familiale. Le nombre de répétitions peut influencer l'âge d'apparition des symptômes et la sévérité de la maladie. Plus le nombre de répétitions est élevé, plus les symptômes sont susceptibles d'être précoces et graves.
Il est important de noter que les répétitions trinucléotides peuvent également être instables pendant la reproduction, ce qui signifie qu'elles ont tendance à s'allonger au fil des générations. Cela peut entraîner une anticipation génétique, où chaque nouvelle génération présente des symptômes plus précoces et plus sévères de la maladie.
En résumé, les répétitions trinucléotides sont des mutations génétiques anormales dans lesquelles une séquence de trois nucléotides est répétée de manière excessive dans un gène donné, ce qui peut entraîner des maladies neurodégénératives héréditaires.
Le cervelet est une structure du système nerveux central située dans la région postérieure du crâne, juste au-dessus du tronc cérébral. Il joue un rôle crucial dans l'intégration et la coordination des mouvements volontaires, ainsi que dans le maintien de l'équilibre et de la posture.
Le cervelet est divisé en deux hémisphères latéraux et une région médiane appelée le vermis. Il contient des neurones spécialisés appelés cellules de Purkinje, qui sont responsables du traitement des informations sensorielles et de la coordination des mouvements musculaires.
Les afférences sensorielles au cervelet proviennent principalement des récepteurs proprioceptifs situés dans les muscles, les tendons et les articulations, ainsi que des informations visuelles et auditives. Le cervelet utilise ces informations pour réguler la force, la direction et la précision des mouvements musculaires, en particulier ceux qui nécessitent une grande coordination et une fine motricité.
Le cervelet est également impliqué dans d'autres fonctions cognitives telles que l'apprentissage moteur, l'attention et la mémoire à court terme. Les dommages au cervelet peuvent entraîner des troubles de l'équilibre, des mouvements anormaux, une dysarthrie (difficulté à articuler les mots), une ataxie (perte de coordination musculaire) et d'autres symptômes neurologiques.
Les protéines gènes suppresseurs de tumeurs sont des protéines qui jouent un rôle crucial dans la régulation du cycle cellulaire et la prévention de la croissance cellulaire anormale. Elles aident à prévenir la transformation des cellules normales en cellules cancéreuses en supprimant l'activation des gènes responsables de la division et de la prolifération cellulaires.
Les gènes qui codent pour ces protéines suppressives de tumeurs sont souvent appelés "gènes suppresseurs de tumeurs". Lorsque ces gènes sont mutés ou endommagés, ils peuvent perdre leur capacité à produire des protéines fonctionnelles, ce qui peut entraîner une augmentation de la division cellulaire et de la croissance tumorale.
Les exemples bien connus de gènes suppresseurs de tumeurs comprennent le gène TP53, qui code pour la protéine p53, et le gène RB1, qui code pour la protéine Rb. Les mutations de ces gènes sont associées à un risque accru de développer certains types de cancer.
En résumé, les protéines gènes suppresseurs de tumeurs sont des protéines qui aident à réguler la croissance cellulaire et à prévenir la formation de tumeurs en supprimant l'activation des gènes responsables de la division et de la prolifération cellulaires.
Les protéines du cycle cellulaire sont des protéines régulatrices clés qui contrôlent et coordonnent les étapes critiques du cycle cellulaire, qui est le processus ordonné par lequel une cellule se divise en deux cellules identiques. Le cycle cellulaire consiste en quatre phases principales: la phase G1 (gap 1), la phase S (synthesis ou synthèse de l'ADN), la phase G2 (gap 2) et la mitose (qui comprend la prophase, la métaphase, l'anaphase et la télophase), suivie de la cytokinèse pour séparer les deux cellules.
Les protéines du cycle cellulaire comprennent des kinases cycline-dépendantes (CDK) et leurs inhibiteurs associés, qui régulent l'entrée dans la phase S et la progression de la mitose. Les cyclines sont des protéines régulatrices qui se lient aux CDK pour activer les complexes kinase CDK-cycline. L'activité des CDK est également régulée par des modifications post-traductionnelles telles que la phosphorylation et la déphosphorylation, ainsi que par la localisation subcellulaire.
Les protéines du cycle cellulaire jouent un rôle essentiel dans le maintien de l'intégrité génomique en coordonnant les événements du cycle cellulaire avec la réparation de l'ADN et la réponse aux dommages à l'ADN. Les dysfonctionnements des protéines du cycle cellulaire peuvent entraîner une régulation anormale du cycle cellulaire, ce qui peut conduire au développement de maladies telles que le cancer.
Les cellules de Purkinje sont un type de neurones situés dans la couche profonde du cervelet, une structure située à l'arrière du cerveau. Elles forment des arbres dendritiques complexes et reçoivent des informations afférentes de plusieurs sources différentes, y compris les noyaux vestibulaires, les noyaux profonds du cervelet et les fibres climbing (qui grimpent le long des dendrites des cellules de Purkinje).
Les cellules de Purkinje sont considérées comme les principales unités fonctionnelles du cervelet, car elles intègrent et traitent les informations entrantes pour produire une sortie vers les noyaux profonds du cervelet. Les signaux de sortie des cellules de Purkinje inhibent les neurones des noyaux profonds, ce qui permet au cervelet de réguler la motricité et d'autres fonctions telles que le contrôle cognitif et l'émotion.
Les cellules de Purkinje sont caractérisées par leur grande taille, avec un diamètre allant jusqu'à 50 micromètres, et par la présence de nombreuses expansions dendritiques couvertes de épines dendritiques. Les épines dendritiques reçoivent des synapses excitatrices de fibres climbing et d'autres afférences, tandis que le soma et les segments initiaux des axones reçoivent des synapses inhibitrices de neurones interneuronaux.
Les cellules de Purkinje sont également connues pour leur vulnérabilité à la dégénérescence dans certaines maladies neurologiques, telles que les ataxies héréditaires et la maladie de Huntington. La perte de ces neurones peut entraîner des troubles moteurs et cognitifs importants.
En termes médicaux, la généalogie est l'étude systématique des antécédents familiaux et médicaux d'une personne ou d'une famille sur plusieurs générations. Elle vise à identifier les modèles de maladies héréditaires ou génétiques dans une famille, ce qui peut aider à évaluer le risque de développer certaines affections et à mettre en œuvre des stratégies de prévention et de dépistage appropriées.
Les professionnels de la santé utilisent souvent des arbres généalogiques pour représenter visuellement les relations familiales et les antécédents médicaux. Ces outils peuvent être particulièrement utiles dans la pratique clinique, en particulier lorsqu'il s'agit de maladies rares ou complexes qui ont tendance à se produire dans certaines familles en raison de facteurs génétiques sous-jacents.
En plus d'être un outil important pour la médecine préventive, la généalogie peut également fournir des informations précieuses sur l'histoire naturelle de diverses maladies et conditions, ce qui contribue à faire progresser notre compréhension globale de la pathogenèse et de la physiopathologie. Par conséquent, elle joue un rôle crucial dans la recherche médicale et les soins cliniques.
Les protéines-sérine-thréonine kinases (PSTK) forment une vaste famille d'enzymes qui jouent un rôle crucial dans la régulation des processus cellulaires, tels que la transcription, la traduction, la réparation de l'ADN, la prolifération et la mort cellulaire. Elles sont appelées ainsi en raison de leur capacité à ajouter un groupe phosphate à des résidus de sérine et de thréonine spécifiques sur les protéines, ce qui entraîne un changement dans la structure et la fonction de ces protéines. Ces kinases sont essentielles au bon fonctionnement de la cellule et sont souvent impliquées dans divers processus pathologiques, y compris le cancer, lorsqu'elles sont surexprimées ou mutées.
Les maladies du cervelet sont un groupe divers de conditions qui affectent le fonctionnement et la structure du cervelet. Le cervelet est une partie importante du cerveau qui joue un rôle crucial dans le contrôle des mouvements musculaires, l'équilibre et la coordination. Les maladies du cervelet peuvent entraîner une variété de symptômes, y compris des mouvements anormaux, une mauvaise coordination, une instabilité, une tremblement de repos, une dysarthrie (parole difficile), des nausées, des vertiges et des problèmes cognitifs.
Les maladies du cervelet peuvent être causées par une variété de facteurs, y compris des anomalies génétiques, des infections, des traumatismes crâniens, des tumeurs, des accidents vasculaires cérébraux et d'autres affections médicales. Certaines maladies du cervelet sont présentes à la naissance ou se développent pendant l'enfance, tandis que d'autres peuvent se développer à tout moment de la vie.
Les exemples courants de maladies du cervelet comprennent l'ataxie cérébelleuse, la sclérose en plaques, la maladie de Parkinson, la maladie de Huntington, les tumeurs cérébrales et les accidents vasculaires cérébraux. Le traitement des maladies du cervelet dépend de la cause sous-jacente et peut inclure une combinaison de médicaments, de thérapies de réadaptation, de chirurgie et de soins de soutien.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
Les troubles de la motilité oculaire, également connus sous le nom de nystagmus ou de strabisme, sont des conditions médicales qui affectent le mouvement et l'alignement des yeux. Le nystagmus se réfère à des mouvements involontaires et rythmiques des yeux, qui peuvent être horizontaux, verticaux ou rotatoires. Ces mouvements peuvent entraîner une vision floue ou double, ainsi que des difficultés de concentration et de coordination.
Le strabisme, d'autre part, est une condition dans laquelle les yeux ne sont pas alignés correctement, ce qui peut entraîner une vision double ou une perte de la vision binoculaire stéréoscopique. Les personnes atteintes de strabisme peuvent avoir un œil qui dévie vers l'intérieur (esotropia), vers l'extérieur (exotropia), vers le haut (hypertropia) ou vers le bas (hypotropia).
Ces troubles peuvent être présents à la naissance ou se développer plus tard dans la vie en raison de divers facteurs, tels que des problèmes neurologiques, des lésions cérébrales, des infections, des traumatismes ou des troubles génétiques. Le traitement dépend du type et de la gravité de la condition et peut inclure des lunettes, des exercices oculaires, des patchs, des médicaments ou une chirurgie.
Les protéines du tissu nerveux sont des types spécifiques de protéines qui se trouvent dans les neurones et le tissu nerveux périphérique. Elles jouent un rôle crucial dans la structure, la fonction et la régulation des cellules nerveuses. Parmi les protéines du tissu nerveux les plus importantes, on peut citer:
1. Neurofilaments: Ces protéines forment une partie importante de la structure interne des neurones et aident à maintenir leur intégrité structurelle. Elles sont également utilisées comme marqueurs pour diagnostiquer certaines maladies neurodégénératives.
2. Neurotransmetteurs: Ces protéines sont responsables de la transmission des signaux chimiques entre les neurones. Les exemples incluent la sérotonine, la dopamine et l'acétylcholine.
3. Canaux ioniques: Ces protéines régulent le flux d'ions à travers la membrane cellulaire des neurones, ce qui est essentiel pour la génération et la transmission des impulsions nerveuses.
4. Protéines d'adhésion: Elles aident à maintenir les contacts entre les neurones et d'autres types de cellules dans le tissu nerveux.
5. Enzymes: Les protéines enzymatiques sont importantes pour la régulation des processus métaboliques dans les neurones, y compris la synthèse et la dégradation des neurotransmetteurs.
6. Chaperons moléculaires: Ces protéines aident à plier et à assembler d'autres protéines dans les neurones, ce qui est essentiel pour leur fonction et leur survie.
7. Protéines de structure: Elles fournissent une structure et un soutien aux cellules nerveuses, telles que la tubuline, qui forme des microtubules dans le cytosquelette des neurones.
Des anomalies dans les protéines du tissu nerveux peuvent entraîner divers troubles neurologiques, y compris des maladies neurodégénératives telles que la maladie d'Alzheimer et la maladie de Parkinson.
Le syndrome du chromosome X fragile (SCXF) est un trouble génétique causé par une mutation du gène FMR1 sur le chromosome X. Cette région du chromosome est particulièrement sujette aux ruptures, d'où le nom de "fragile" associé à ce syndrome.
La mutation responsable du SCXF entraîne une expansion répétitive de la séquence CGG dans le gène FMR1. Plus le nombre de répétitions est élevé, plus les symptômes sont graves. Chez les personnes atteintes d'une forme prémutation du SCXF, il y a entre 55 et 200 répétitions de CGG. Ces individus peuvent ne présenter aucun symptôme ou développer des problèmes de santé plus tard dans la vie, tels que des troubles neurologiques ou reproductifs.
Chez les personnes atteintes d'une forme à part entière du SCXF, il y a plus de 200 répétitions de CGG. Cela provoque une méthylation excessive de la région et une transcription réduite du gène FMR1, ce qui entraîne l'absence ou la diminution de la protéine FMRP (protéine associée au syndrome X fragile). Cette protéine joue un rôle crucial dans le développement et le fonctionnement du cerveau.
Les symptômes du SCXF peuvent varier considérablement, allant de légers à sévères. Les manifestations courantes comprennent des retards intellectuels ou du développement, des problèmes d'apprentissage, des troubles du comportement (tels que l'hyperactivité et l'anxiété), des caractéristiques physiques distinctives (comme un visage allongé, de grandes oreilles et une mâchoire proéminente) et des problèmes de santé courants (comme des problèmes cardiaques et articulaires). Les femmes sont souvent moins touchées que les hommes en raison de l'effet protecteur d'un chromosome X supplémentaire.
Le SCXF est généralement héréditaire, transmis par la mère dans un mode lié à l'X. Cependant, environ 30 % des cas sont causés par une nouvelle mutation et ne présentent aucun antécédent familial de la maladie. Il n'existe actuellement aucun traitement pour le SCXF, mais des interventions peuvent être mises en place pour gérer les symptômes et améliorer la qualité de vie des personnes atteintes.
Une apraxie est un trouble du mouvement caractérisé par l'incapacité d'effectuer des mouvements volontaires ou des séquences de mouvements complexes, malgré un contrôle musculaire et cognitif préservé. Ce trouble est généralement causé par une lésion cérébrale, le plus souvent dans les zones du cerveau responsables du traitement et de la planification des mouvements volontaires, telles que les lobes frontaux ou pariétaux.
Les apraxies peuvent affecter différents types de mouvements, tels que la coordination des membres (appelée apraxie motrice), l'utilisation d'outils et d'objets (apraxie idéomotrice), la parole (apraxie verbale) ou les expressions faciales et les gestes sociaux (apraxie bucco-faciale). Les symptômes peuvent inclure des mouvements maladroits, lents, imprécis ou inappropriés, ainsi qu'une difficulté à initier, arrêter ou modifier des mouvements volontaires.
Le diagnostic d'apraxie repose sur une évaluation neurologique approfondie, comprenant un examen physique et neuropsychologique détaillé. Le traitement peut inclure la réadaptation et la thérapie des mouvements, ainsi que l'utilisation d'aides techniques pour faciliter les activités quotidiennes. Dans certains cas, des médicaments peuvent être prescrits pour aider à gérer les symptômes moteurs associés à l'apraxie.
L'âge d'apparition, également connu sous le nom d'âge de début ou d'âge de survenue, fait référence à l'âge auquel une personne développe pour la première fois les symptômes ou manifestations d'une maladie, d'un trouble de santé mentale, d'un handicap ou d'autres conditions médicales. Il peut être exprimé en années, mois ou même semaines après la naissance, selon le type et la gravité de la condition concernée.
L'âge d'apparition est un aspect important du diagnostic et de la prise en charge médicale, car il peut fournir des indices sur les causes sous-jacentes de la maladie, influencer le choix des traitements et des interventions, et aider à prévoir l'évolution et le pronostic de la condition.
Par exemple, certaines maladies génétiques ou congénitales peuvent se manifester dès la naissance ou dans les premiers mois de vie, tandis que d'autres troubles, tels que la maladie d'Alzheimer ou la démence, ne se développent généralement qu'à un âge plus avancé. De même, certains troubles mentaux, comme l'autisme et le trouble déficitaire de l'attention avec hyperactivité (TDAH), peuvent présenter des signes avant-coureurs dès la petite enfance, tandis que d'autres, comme la schizophrénie, peuvent ne se manifester qu'à l'adolescence ou à l'âge adulte.
Il est important de noter que l'âge d'apparition peut varier considérablement d'une personne à l'autre, même au sein d'une même famille ou d'un même groupe de population. Des facteurs tels que les antécédents familiaux, l'environnement, le mode de vie et d'autres facteurs de risque peuvent influencer le moment où une personne développe les symptômes d'une condition donnée.
La dysarthrie est un trouble de la parole dû à une lésion cérébrale ou neurologique qui affecte les muscles utilisés dans la production de la parole. Cela peut entraîner une variété de symptômes, y compris une articulation imprécise, une intonation et un rythme anormaux, une voix faible ou rauque, et une salivaire excessive pendant la parole. La sévérité de ces symptômes peut varier considérablement, allant d'une légère difficulté à être compris à une incapacité complète de produire des mots intelligibles. Les causes courantes de dysarthrie comprennent les accidents vasculaires cérébraux, les traumatismes crâniens, les tumeurs cérébrales, certaines maladies neurodégénératives telles que la sclérose en plaques et la maladie de Parkinson. Le traitement de la dysarthrie dépend généralement de l'identification et du traitement de la cause sous-jacente, ainsi que de la thérapie de la parole pour aider à améliorer la fonction de la parole.
Les protéines fixant l'ADN, également connues sous le nom de protéines liant l'ADN ou protéines nucléaires, sont des protéines qui se lient spécifiquement à l'acide désoxyribonucléique (ADN). Elles jouent un rôle crucial dans la régulation de la transcription et de la réplication de l'ADN, ainsi que dans la maintenance de l'intégrité du génome.
Les protéines fixant l'ADN se lient à des séquences d'ADN spécifiques grâce à des domaines de liaison à l'ADN qui reconnaissent et se lient à des motifs particuliers dans la structure de l'ADN. Ces protéines peuvent agir comme facteurs de transcription, aidant à activer ou à réprimer la transcription des gènes en régulant l'accès des polymérases à l'ADN. Elles peuvent également jouer un rôle dans la réparation de l'ADN, en facilitant la reconnaissance et la réparation des dommages à l'ADN.
Les protéines fixant l'ADN sont souvent régulées elles-mêmes par des mécanismes post-traductionnels tels que la phosphorylation, la méthylation ou l'acétylation, ce qui permet de moduler leur activité en fonction des besoins cellulaires. Des anomalies dans les protéines fixant l'ADN peuvent entraîner diverses maladies génétiques et sont souvent associées au cancer.
La protéine du syndrome X fragile, également connue sous le nom de protéine FMR1, est une protéine qui joue un rôle crucial dans la structure et la fonction des synapses, qui sont les sites de communication entre les neurones dans le cerveau. Cette protéine est codée par le gène FMR1 situé sur le chromosome X.
Dans le syndrome X fragile, qui est la forme héréditaire la plus courante de handicap intellectuel, une expansion répétitive de la séquence de triplets CGG dans le gène FMR1 entraîne une méthylation de l'ADN et une transcription réduite ou absente du gène, ce qui entraîne une absence ou une diminution significative de la protéine FMR1. Cette perte de fonction est associée à une gamme de symptômes, notamment des retards de développement, des déficiences intellectuelles, des troubles du comportement et des anomalies physiques.
La protéine FMR1 régule également l'expression d'autres gènes et joue un rôle important dans la plasticité synaptique, qui est la capacité des synapses à se renforcer ou à s'affaiblir en réponse aux stimuli. Par conséquent, une altération de la protéine FMR1 peut avoir des effets dévastateurs sur le développement et la fonction du cerveau.
Un gène récessif est un type de gène qui doit être présent en double exemplaire (un hérité de chaque parent) pour qu'un trait ou une condition particulière se manifeste chez un individu. Dans l'espèce humaine, les caractéristiques contrôlées par des gènes récessifs ne s'expriment que si un individu hérite d'une copie de ce gène récessif de chaque parent.
Si un seul parent transmet le gène récessif, l'individu sera quand même porteur du trait récessif mais ne le manifestera pas, on l'appelle alors un porteur sain. Ce n'est que lorsque deux personnes qui sont toutes les deux porteuses d'un gène récessif ont un enfant qu'il y a une chance sur quatre (25%) à chaque grossesse pour que l'enfant hérite du trait récessif des deux parents et manifeste donc la caractéristique ou la maladie associée à ce gène.
Un exemple couramment cité de gène récessif est celui responsable de la drépanocytose, une maladie génétique affectant les globules rouges. Pour développer cette maladie, un individu doit hériter d'une copie du gène anormal de chaque parent. Si l'individu n'en hérite que d'une seule copie, il sera porteur sain du trait drépanocytaire mais ne développera pas la maladie.
Un syndrome, dans le contexte médical, est un ensemble de symptômes ou de signes cliniques qui, considérés dans leur globalité, suggèrent l'existence d'une pathologie spécifique ou d'un état anormal dans le fonctionnement de l'organisme. Il s'agit essentiellement d'un ensemble de manifestations cliniques qui sont associées à une cause sous-jacente commune, qu'elle soit connue ou inconnue.
Un syndrome n'est pas une maladie en soi, mais plutôt un regroupement de signes et symptômes qui peuvent être liés à différentes affections médicales. Par exemple, le syndrome métabolique est un ensemble de facteurs de risque qui augmentent la probabilité de développer des maladies cardiovasculaires et du diabète de type 2. Ces facteurs comprennent l'obésité abdominale, l'hypertension artérielle, l'hyperglycémie à jeun et les taux élevés de triglycérides et de faibles taux de HDL-cholestérol.
La définition d'un syndrome peut évoluer avec le temps, alors que la compréhension des mécanismes sous-jacents s'améliore grâce aux recherches médicales et scientifiques. Certains syndromes peuvent être nommés d'après les professionnels de la santé qui ont contribué à leur identification ou à leur description, comme le syndrome de Down (trisomie 21) ou le syndrome de Klinefelter (XXY).
Il est important de noter que la présence d'un syndrome ne permet pas toujours d'établir un diagnostic définitif, car plusieurs affections médicales peuvent partager des symptômes similaires. Cependant, l'identification d'un syndrome peut aider les professionnels de la santé à orienter le diagnostic et le traitement vers des causes probables ou à fournir des informations sur le pronostic et la prise en charge globale du patient.
Les lésions de l'ADN se réfèrent à toute modification ou dommage dans la structure de l'acide désoxyribonucléique (ADN), qui est le matériel génétique présent dans les cellules. Ces lésions peuvent être causées par divers facteurs, tels que les radiations ionisantes, les produits chimiques toxiques, les erreurs de réplication de l'ADN, certains médicaments et agents infectieux, ainsi que les processus naturels du vieillissement.
Les lésions de l'ADN peuvent entraîner des mutations génétiques, qui peuvent altérer la fonction des gènes et contribuer au développement de maladies telles que le cancer. Les dommages à l'ADN peuvent également déclencher des mécanismes de réparation de l'ADN, qui sont essentiels pour maintenir l'intégrité du génome. Cependant, si les lésions sont trop graves ou ne sont pas correctement réparées, elles peuvent entraîner une mort cellulaire programmée (apoptose) ou la transformation maligne des cellules.
Les types courants de lésions de l'ADN comprennent les cassures simples et doubles brins, les dimères de thymine, les oxydations de bases, les modifications chimiques des bases, les adduits de base et les cross-links entre les deux brins d'ADN. Des tests spécifiques peuvent être utilisés pour détecter et évaluer ces lésions dans le cadre du diagnostic et du traitement de diverses maladies.
La dyssynergie cérébelleuse myoclonique est un trouble du mouvement caractérisé par des contractions musculaires brusques et incontrôlables (myoclonies) associées à une mauvaise coordination musculaire (dyssynergie) et à une instabilité posturale. Ces symptômes sont dus à une dysfonction du cervelet, une structure du cerveau responsable de la coordination des mouvements volontaires.
Les myoclonies peuvent affecter un muscle ou un groupe de muscles et peuvent être déclenchées par des mouvements volontaires, des réflexes ou des stimulations sensorielles telles que des bruits soudains ou des touches légères. Les personnes atteintes de dyssynergie cérébelleuse myoclonique peuvent également présenter une variété d'autres symptômes tels qu'une démarche instable, des mouvements oculaires anormaux, une dysarthrie (parole difficile), une dysphagie (difficulté à avaler) et une ataxie (perte de coordination des mouvements volontaires).
La dyssynergie cérébelleuse myoclonique peut être causée par divers facteurs, notamment des lésions cérébrales, des maladies dégénératives du système nerveux central, des infections, des intoxications et des troubles métaboliques. Dans certains cas, la cause de la dyssynergie cérébelleuse myoclonique est inconnue, ce qui est alors qualifié d'idiopathique.
Le traitement de la dyssynergie cérébelleuse myoclonique dépend de la cause sous-jacente et peut inclure des médicaments anticonvulsivants, des agents antiépileptiques, des benzodiazépines ou d'autres thérapies. La physiothérapie et l'ergothérapie peuvent également être bénéfiques pour améliorer la fonction musculaire et la coordination.
Le nystagmus pathologique est un mouvement involontaire et oscillatoire des yeux qui peut être dû à une variété de conditions médicales sous-jacentes affectant le système nerveux central ou périphérique. Ce trouble de la fixation visuelle se caractérise par des mouvements rapides et lents des yeux dans une direction donnée, suivis d'un mouvement réflexe dans la direction opposée.
Le nystagmus pathologique peut être horizontal, vertical ou rotatoire (torsionnel) en fonction de la cause sous-jacente. Il peut être congénital ou acquis et peut entraîner une vision floue, une instabilité posturale et des maux de tête. Les causes courantes du nystagmus pathologique comprennent les lésions cérébrales, les tumeurs cérébrales, les infections de l'oreille interne, les troubles vestibulaires, les intoxications, les traumatismes crâniens et certaines maladies neurologiques dégénératives.
Le traitement du nystagmus pathologique dépend de la cause sous-jacente et peut inclure des médicaments, une intervention chirurgicale ou des thérapies de réadaptation visuelle. Dans certains cas, le nystagmus peut être difficile à traiter et peut entraîner une déficience visuelle permanente.
L'expansion de séquences répétées d'ADN est un phénomène génétique où le nombre de répétitions d'une certaine séquence nucléotidique particulière dans une région spécifique d'un gène s'allonge de manière anormale. Ce processus se produit généralement pendant la réplication de l'ADN et peut être influencé par des facteurs tels que l'âge, la fonction réparatrice de l'ADN et d'autres facteurs environnementaux.
Les séquences répétées d'ADN sont courantes dans le génome humain, mais lorsqu'elles sont répétées un certain nombre de fois, elles peuvent entraîner des modifications de la structure et de la fonction des gènes. Dans l'expansion de séquences répétées d'ADN, le nombre de répétitions peut atteindre plusieurs centaines ou même milliers, ce qui provoque une instabilité génétique et perturbe la fonction normale du gène.
Cette expansion de séquences répétées d'ADN est associée à un certain nombre de maladies neurodégénératives et neuromusculaires héréditaires, telles que la maladie de Huntington, la maladie de Charcot-Marie-Tooth, la sclérose latérale amyotrophique (SLA) et l'ataxie spinocérébelleuse. Les symptômes et la gravité de ces maladies dépendent souvent du nombre de répétitions dans le gène affecté.
Le canal potassique Kv1.1 est un type spécifique de canal ionique qui permet le mouvement des ions potassium à travers la membrane cellulaire. Il s'agit d'un canal voltage-dépendant, ce qui signifie que son ouverture et sa fermeture sont régulées par les changements du potentiel électrique de la membrane.
Le gène KCNA1 code pour le canal potassique Kv1.1. Les mutations de ce gène peuvent entraîner des maladies neuromusculaires, telles que le syndrome de l'X fragile associé au nodopectinal et l'épilepsie familiale bénigne du type 2 (BFES2).
Le canal potassique Kv1.1 joue un rôle important dans la génération et la propagation des impulsions nerveuses, ainsi que dans le maintien de l'excitabilité neuronale. Il est particulièrement abondant dans les nœuds de Ranvier des fibres nerveuses périphériques, où il contribue à la formation du potentiel d'action localisé et à la transmission rapide des impulsions nerveuses.
Une remnographie est un type d'examen d'imagerie médicale qui utilise une faible dose de radiation pour produire des images détaillées des structures internes du corps. Contrairement à une radiographie standard, une remnographie implique l'utilisation d'un milieu de contraste, comme un produit de contraste à base d'iode, qui est ingéré ou injecté dans le patient avant l'examen.
Le milieu de contraste permet aux structures internes du corps, telles que les vaisseaux sanguins, les organes creux ou les tissus mous, d'être plus visibles sur les images radiographiques. Cela peut aider les médecins à diagnostiquer une variété de conditions médicales, y compris les maladies gastro-intestinales, les maladies rénales et les troubles vasculaires.
Les remnographies sont généralement considérées comme sûres, bien que comme avec toute procédure médicale qui utilise des radiations, il existe un risque minimal de dommages aux tissus ou au matériel génétique. Les avantages potentiels d'un diagnostic précis et opportun sont généralement considérés comme dépassant ce faible risque.
Il est important de noter que les remnographies ne doivent être effectuées que lorsqu'elles sont médicalement nécessaires, car l'exposition répétée aux radiations peut augmenter le risque de dommages à long terme. Les médecins et les technologues en imagerie médicale prennent des précautions pour minimiser l'exposition aux radiations pendant les procédures de remnographie.
Le phénotype est le résultat observable de l'expression des gènes en interaction avec l'environnement et d'autres facteurs. Il s'agit essentiellement des manifestations physiques, biochimiques ou développementales d'un génotype particulier.
Dans un contexte médical, le phénotype peut se rapporter à n'importe quelle caractéristique mesurable ou observable résultant de l'interaction entre les gènes et l'environnement, y compris la couleur des yeux, la taille, le poids, certaines maladies ou conditions médicales, voire même la réponse à un traitement spécifique.
Il est important de noter que deux individus ayant le même génotype (c'est-à-dire la même séquence d'ADN) ne seront pas nécessairement identiques dans leur phénotype, car des facteurs environnementaux peuvent influencer l'expression des gènes. De même, des individus avec des génotypes différents peuvent partager certains traits phénotypiques en raison de similitudes dans leurs environnements ou dans d'autres facteurs non génétiques.
Les mutants neurologiques de souris sont des souches de rongeurs qui présentent des anomalies ou des modifications génétiques spécifiques dans leur système nerveux central et périphérique. Ces mutations peuvent entraîner une variété de phénotypes, tels que des troubles de l'apprentissage, de la mémoire, du mouvement, de la coordination, de l'humeur ou de la sensibilité sensorielle.
Les mutants neurologiques de souris sont souvent utilisés comme modèles animaux dans la recherche biomédicale pour étudier les mécanismes sous-jacents des maladies humaines du système nerveux, telles que les troubles neurodégénératifs (maladie d'Alzheimer, Parkinson), les troubles psychiatriques (schizophrénie, dépression) et les troubles neurologiques développementaux (autisme, épilepsie).
Les mutations peuvent affecter des gènes spécifiques ou des voies de signalisation, ce qui permet aux chercheurs d'étudier l'impact de ces modifications sur le fonctionnement du cerveau et du système nerveux. Ces modèles animaux sont essentiels pour comprendre les bases moléculaires et cellulaires des maladies neurologiques et pour tester de nouvelles thérapies et traitements potentiels.
Les protéines nucléaires sont des protéines qui se trouvent dans le noyau des cellules et jouent un rôle crucial dans la régulation de l'expression des gènes, la réplication de l'ADN, la réparation de l'ADN, la transcription de l'ARN et d'autres processus essentiels à la survie et à la reproduction des cellules.
Il existe plusieurs types de protéines nucléaires, y compris les histones, qui sont des protéines structurelles qui aident à compacter l'ADN en chromosomes, et les facteurs de transcription, qui se lient à l'ADN pour réguler l'expression des gènes. Les protéines nucléaires peuvent également inclure des enzymes qui sont impliquées dans la réplication et la réparation de l'ADN, ainsi que des protéines qui aident à maintenir l'intégrité structurelle du noyau.
Les protéines nucléaires peuvent être régulées au niveau de leur expression, de leur localisation dans la cellule et de leur activité enzymatique. Des anomalies dans les protéines nucléaires peuvent entraîner des maladies génétiques et contribuer au développement du cancer. Par conséquent, l'étude des protéines nucléaires est importante pour comprendre les mécanismes moléculaires qui sous-tendent la régulation de l'expression des gènes et d'autres processus cellulaires essentiels.
Un corps d'inclusion intranucléaire est une structure sphérique ou ovoïde, fortement basophile (qui a une affinité pour les colorants basiques), observée dans le noyau de certaines cellules. Il s'agit d'une inclusion pathologique, souvent liée à diverses maladies neurodégénératives et virales. Les corps d'inclusion intranucléaires sont composés de protéines anormalement accumulées et/ou de matériel génétique, qui peuvent entraver les fonctions cellulaires normales et contribuer à la dégénérescence des neurones. Des exemples de ces conditions incluent certaines formes de démences, comme la maladie de Creutzfeldt-Jakob, et certains types d'infections virales, telles que les encéphalites.
Les atrophies olivo-ponto-cérébelleuses sont un groupe de troubles neurologiques caractérisés par la dégénérescence et l'atrophie des structures cérébrales, y compris le cervelet, le pont et les noyaux olivaires. Ces troubles peuvent être héréditaires ou sporadiques et se manifestent généralement par une combinaison de symptômes tels que des mouvements anormaux, une coordination altérée, une instabilité, une dysarthrie (parole difficile), une nystagmus (mouvement involontaire des yeux) et une ataxie (perte de coordination musculaire).
Les causes sous-jacentes des atrophies olivo-ponto-cérébelleuses peuvent varier, mais comprennent souvent des mutations génétiques qui affectent la fonction des neurones. Les symptômes peuvent apparaître à tout âge, selon le type de trouble, et peuvent s'aggraver progressivement au fil du temps.
Le diagnostic des atrophies olivo-ponto-cérébelleuses est généralement posé sur la base d'un examen neurologique approfondi, y compris une évaluation des mouvements et de l'équilibre, ainsi que d'une imagerie cérébrale pour confirmer l'atrophie des structures cérébelleuses. Le traitement est généralement symptomatique et peut inclure des médicaments pour aider à contrôler les mouvements anormaux, la physiothérapie pour améliorer la force et la coordination musculaires, et des modifications du mode de vie pour faciliter les activités quotidiennes.
Un gène dominant est un type de gène qui, lorsqu'il est exprimé, sera manifestement exprimé dans l'organisme, que le gène ait deux copies (hétérozygote) ou deux copies identiques (homozygote). Cela signifie qu'une seule copie du gène dominant est suffisante pour provoquer une certaine apparence phénotypique ou une condition médicale, qui peut être bénigne ou pathologique.
Dans le contexte de la génétique mendélienne classique, un trait dominant est exprimé dans la progéniture même si l'autre copie du gène est normale (saine). Les traits dominants se manifestent généralement dans chaque génération et sont plus susceptibles d'être observés dans les familles affectées.
Un exemple bien connu de gène dominant est le gène de la névoid basocellulaire syndrome (NBS), également appelé syndrome du grain de beauté multiple, qui prédispose les individus à développer des cancers de la peau. Si un parent a ce trait et ne possède qu'une seule copie du gène NBS muté, chaque enfant aura 50% de chances d'hériter de cette copie mutée et donc de développer le syndrome.
Les maladies neurodégénératives héréditaires sont un groupe de troubles du système nerveux central qui entraînent une détérioration progressive et irréversible des neurones ou cellules nerveuses. Ces maladies sont dites «héréditaires» car elles sont liées à des mutations génétiques spécifiques qui peuvent être transmises de génération en génération.
Ces affections se caractérisent par une perte progressive des fonctions cognitives, motrices ou sensorielles. Elles incluent des maladies telles que la maladie de Huntington, certaines formes de sclérose latérale amyotrophique (SLA), la maladie de Parkinson à début précoce, et certaines formes de démence familiale comme la maladie de Alzheimer à début précoce.
La cause sous-jacente est une anomalie dans les gènes qui codent pour des protéines spécifiques. Ces protéines anormales s'accumulent dans les neurones, entraînant leur dysfonctionnement et leur mort. Cela conduit à la dégénérescence du tissu cérébral et à une variété de symptômes neurologiques.
Il est important de noter que tous les cas de maladies neurodégénératives ne sont pas héréditaires ; certaines sont dites «sporadiques», ce qui signifie qu'elles ne semblent pas être liées à des facteurs génétiques spécifiques. Cependant, même dans ces cas, il peut y avoir une composante génétique qui influence le risque de développer la maladie.
Une mutation « faux sens » (ou missense mutation) est un type de mutation génétique où une seule paire de bases dans l'ADN est modifiée, ce qui entraîne le remplacement d'un acide aminé par un autre dans la protéine codée par ce gène. Cela peut altérer la fonction, la structure ou la stabilité de la protéine, dépendant de la position et de l'importance de l'acide aminé remplacé. Dans certains cas, ces mutations peuvent entraîner des maladies génétiques ou prédisposer à certaines conditions médicales. Toutefois, il est important de noter que toutes les mutations faux sens ne sont pas nécessairement pathogènes et que leur impact sur la santé dépend du contexte dans lequel elles se produisent.
L'atrophie multisystématisée (AMS) est une maladie neurodégénérative rare qui affecte plusieurs systèmes du corps, y compris le système nerveux central et périphérique. Elle se caractérise par la dégénération et la perte de neurones dans différentes régions du cerveau, entraînant une variété de symptômes.
Les symptômes les plus courants de l'AMS comprennent :
* La rigidité musculaire et les tremblements, similaires à la maladie de Parkinson ;
* La perte d'équilibre et de coordination, semblable à l'atrophie musculaire progressive ;
* Les problèmes de contrôle de la vessie et de la fonction intestinale ;
* Les troubles cognitifs et émotionnels, tels que la démence et la dépression.
La cause exacte de l'AMS est inconnue, mais on pense qu'elle est liée à une combinaison de facteurs génétiques et environnementaux. Il n'existe actuellement aucun traitement curatif pour l'AMS, bien que certains médicaments puissent aider à soulager les symptômes.
Le diagnostic de l'AMS peut être difficile en raison de la variété des symptômes et de leur chevauchement avec d'autres maladies neurologiques. Les médecins utilisent généralement une combinaison de tests, y compris les antécédents médicaux du patient, l'examen physique, les tests de laboratoire et les techniques d'imagerie cérébrale pour diagnostiquer l'AMS.
L'évolution de la maladie peut varier considérablement d'un individu à l'autre, mais elle est généralement progressive et débilitante. La durée de vie moyenne après le diagnostic est d'environ 6 à 10 ans.
L'encéphale est la structure centrale du système nerveux situé dans la boîte crânienne. Il comprend le cerveau, le cervelet et le tronc cérébral. L'encéphale est responsable de la régulation des fonctions vitales telles que la respiration, la circulation sanguine et la température corporelle, ainsi que des fonctions supérieures telles que la pensée, la mémoire, l'émotion, le langage et la motricité volontaire. Il est protégé par les os de la boîte crânienne et recouvert de trois membranes appelées méninges. Le cerveau et le cervelet sont floating dans le liquide céphalo-rachidien, qui agit comme un coussin pour amortir les chocs et les mouvements brusques.
La gliadine est une protéine que l'on trouve dans le gluten, présent principalement dans les céréales comme le blé, l'orge et le seigle. Elle est composée de plusieurs fractions protéiques, dont certaines sont responsables de la réaction immunitaire anormale chez les personnes atteintes de maladie coeliaque. Cette maladie est une affection auto-immune dans laquelle l'ingestion de gliadine entraîne des dommages à la muqueuse de l'intestin grêle, provoquant ainsi des symptômes gastro-intestinaux et diverses complications systémiques.
Il est important de noter que les personnes atteintes de maladie coeliaque doivent suivre un régime strict sans gluten, ce qui signifie éviter toute consommation de gliadine pour prévenir l'apparition des symptômes et des dommages aux intestins. D'autres affections liées au gluten, telles que la sensibilité au gluten non coeliaque et la dermatite herpétiforme, peuvent également bénéficier d'un régime sans gluten pour gérer les symptômes et améliorer la qualité de vie.
La carence en vitamine E est un état nutritionnel qui se produit lorsqu'un individu ne consomme pas ou ne peut pas absorber suffisamment de vitamine E, un nutriment liposoluble essentiel. La vitamine E est une puissante antioxydante qui protège les cellules du corps contre les dommages causés par les radicaux libres. Les radicaux libres sont des molécules instables qui peuvent endommager les membranes cellulaires, les protéines et l'ADN.
Les symptômes de la carence en vitamine E comprennent une faiblesse musculaire, une perte d'équilibre, une mauvaise coordination, des tremblements, des dommages aux nerfs périphériques et une vision réduite. Dans les cas graves, la carence en vitamine E peut entraîner une maladie neuromusculaire appelée myopathie neurogénique, qui affecte les muscles squelettiques et les nerfs périphériques.
La carence en vitamine E est rare chez les personnes qui suivent un régime alimentaire équilibré et varié. Cependant, certaines personnes peuvent être à risque de carence en vitamine E, notamment celles atteintes de maladies chroniques du foie ou des intestins, de fibrose kystique, de mucoviscidose ou de malabsorption intestinale. Les nouveau-nés prématurés peuvent également être à risque de carence en vitamine E en raison de leur faible stockage de graisse corporelle et de leur immaturité hépatique.
Le traitement de la carence en vitamine E consiste généralement à fournir des suppléments de vitamine E par voie orale ou intraveineuse. Les sources alimentaires riches en vitamine E comprennent les huiles végétales, les noix et les graines, les légumes verts feuillus, les avocats et les poissons gras. Il est important de consulter un médecin avant de prendre des suppléments de vitamine E, car une surdose peut entraîner des effets secondaires indésirables tels que des nausées, des vomissements, des diarrhées et des maux de tête.
Un examen neurologique est un processus systématique d'évaluation des fonctions et structures du système nerveux central (cerveau et moelle épinière) et périphérique (nerfs crâniens, nerfs spinaux et leurs racines, plaque motrice et réflexes). Il est utilisé pour diagnostiquer les troubles neurologiques, suivre la progression de maladies connues ou évaluer l'efficacité du traitement.
L'examen comprend typiquement une série de tests qui visent à évaluer :
1. La conscience et le niveau de vigilance.
2. Les fonctions cognitives telles que la mémoire, le langage, l'orientation dans le temps et l'espace.
3. Les mouvements oculaires, la vision et la perception visuelle.
4. La force musculaire, la coordination et l'équilibre.
5. Les réflexes tendineux profonds et cutanés.
6. La sensibilité à la douleur, au toucher, à la température et aux vibrations.
7. Les fonctions des nerfs crâniens (olfaction, vision, ouïe, goût, mouvements faciaux, déglutition, etc.)
Les résultats de ces tests aident les médecins à identifier les zones du système nerveux qui pourraient être endommagées ou malades, ce qui peut conduire à un diagnostic plus précis et à un plan de traitement approprié.
Les canaux calciques de type-L, également connus sous le nom de canaux calciques voltage-dépendants lents (CVDL), sont des protéines membranaires qui jouent un rôle crucial dans la régulation du calcium intracellulaire dans les cellules excitables telles que les muscles lisses et les cardiomyocytes.
Ils sont appelés "canaux calciques de type-L" en raison de leur activation lente et prolongée en réponse à des changements de potentiel membranaire dépolarisant, ce qui permet une entrée soutenue de calcium dans la cellule. Cette entrée de calcium déclenche ensuite la libération de calcium supplémentaire à partir du réticulum sarcoplasmique, entraînant ainsi la contraction musculaire.
Les mutations dans les gènes qui codent pour ces canaux calciques peuvent entraîner des maladies cardiaques héréditaires telles que le syndrome du QT long, qui se caractérise par une durée anormalement longue de l'intervalle QT sur l'électrocardiogramme (ECG) et un risque accru d'arythmies ventriculaires potentiellement mortelles. D'autres mutations peuvent entraîner une hypertension artérielle ou une insuffisance cardiaque congestive.
Il est important de noter que la régulation adéquate des canaux calciques de type-L est essentielle pour maintenir un rythme cardiaque normal et une fonction musculaire appropriée, et tout dysfonctionnement peut entraîner de graves conséquences sur la santé.
Dans le domaine de la génétique, un individu homozygote est une personne qui hérite de deux allèles identiques pour un trait spécifique, ayant reçu ce même allèle de chacun de ses deux parents. Cela peut se traduire par l'expression d'un caractère particulier ou l'apparition d'une maladie génétique dans le cas où ces allèles sont défectueux.
On distingue deux types d'homozygotes : les homozygotes dominants et les homozygotes récessifs. Les homozygotes dominants présentent le phénotype lié au gène dominant, tandis que les homozygotes récessifs expriment le phénotype associé au gène récessif.
Par exemple, dans le cas de la drépanocytose, une maladie génétique héréditaire affectant l'hémoglobine des globules rouges, un individu homozygote pour cette affection possède deux allèles mutés du gène de l'hémoglobine et exprimera donc les symptômes de la maladie.
La myoclonie est un type de spasme musculaire involontaire caractérisé par des contractions soudaines et brèves des muscles, souvent comparées à des secousses ou des mouvements ressemblant à des battements. Ces mouvements peuvent affecter un seul muscle ou un groupe de muscles et peuvent survenir au repos ou pendant des activités volontaires. Les myoclonies peuvent être causées par divers facteurs, tels que des lésions cérébrales, des maladies neurodégénératives, des infections, des troubles métaboliques ou des réactions médicamenteuses. Elles peuvent également se produire sans cause apparente et sont alors appelées myoclonies essentielles. Dans certains cas, les myoclonies peuvent être traitées avec des médicaments ou d'autres thérapies, mais dans d'autres situations, elles peuvent être difficiles à gérer et avoir un impact significatif sur la qualité de vie d'une personne.
Les canaux calciques de type L, également connus sous le nom de canaux calciques voltage-dépendants lents (CVDL), sont des protéines membranaires qui jouent un rôle crucial dans la régulation du calcium intracellulaire dans les cellules excitables telles que les muscles lisses et cardiaques, ainsi que dans certaines neurones.
Ces canaux sont sensibles aux changements de potentiel membranaire et s'ouvrent en réponse à des dépolarisations membranaires, permettant au calcium de pénétrer dans la cellule. Cette entrée de calcium déclenche une cascade de réactions qui conduisent à la contraction musculaire ou à la transmission de signaux nerveux.
Les mutations génétiques dans les gènes codant pour ces canaux peuvent entraîner des maladies cardiaques héréditaires telles que le syndrome du QT long, qui se caractérise par une prolongation de l'intervalle QT sur l'électrocardiogramme et un risque accru d'arythmies ventriculaires potentiellement mortelles. D'autres mutations peuvent entraîner une hypertension artérielle pulmonaire, une insuffisance cardiaque ou des migraines.
Les canaux calciques de type L sont donc des cibles thérapeutiques importantes pour le traitement de diverses maladies cardiovasculaires et neurologiques.
Les maladies neurodégénératives sont un groupe de conditions médicales progressives qui impliquent une dégénérescence et une mort cellulaire dans les neurones ou les cellules nerveuses du cerveau. Ce processus entraîne des dommages aux connections nerveuses et, finalement, à la perte des fonctions neurologiques essentielles.
Les symptômes de ces maladies varient en fonction de la région du cerveau qui est affectée. Ils peuvent inclure des problèmes cognitifs comme la démence, des problèmes moteurs tels que les tremblements ou la difficulté à bouger, et d'autres problèmes comme la perte de l'odorat ou la dépression.
Les exemples courants de maladies neurodégénératives comprennent la maladie d'Alzheimer, la maladie de Parkinson, la sclérose latérale amyotrophique (SLA), la démence à corps de Lewy, et la maladie de Huntington. Ces maladies sont généralement incurables et les traitements disponibles visent principalement à soulager les symptômes et à améliorer la qualité de vie des patients.
L'atrophie est un terme médical qui décrit la diminution de la taille ou du volume d'un tissu, d'un organe ou d'une partie du corps en raison de la perte de cellules ou de la réduction de leur taille. Cela peut être causé par une variété de facteurs, y compris le vieillissement, les maladies chroniques, l'inactivité physique, la dénutrition et les lésions nerveuses.
Les exemples courants d'atrophie comprennent la fonte musculaire due à l'immobilisation prolongée, la perte de tissu cérébral dans des conditions telles que la maladie d'Alzheimer ou la sclérose en plaques, et la réduction de la taille de la glande mammaire chez les femmes qui allaitent.
Les symptômes de l'atrophie dépendent de la zone du corps affectée. Ils peuvent inclure une faiblesse musculaire, une perte d'équilibre, des mouvements plus lents et moins précis, une diminution de la fonction sensorielle, une modification de la voix ou de la vision, et dans certains cas, des douleurs ou des crampes.
Le traitement de l'atrophie dépend de la cause sous-jacente. Dans certains cas, il peut être possible de ralentir ou d'arrêter le processus d'atrophie en traitant la maladie sous-jacente. Dans d'autres cas, des exercices de renforcement musculaire, une thérapie physique ou occupationnelle, et des changements de mode de vie peuvent aider à améliorer les symptômes et la fonction.
La réparation de l'ADN est le processus biologique par lequel les cellules identifient et corrigent les dommages à l'acide désoxyribonucléique (ADN), qui est le matériel génétique présent dans les chromosomes des cellules. L'ADN peut être endommagé par divers facteurs, y compris les radiations, les produits chimiques et les erreurs de réplication qui se produisent lorsque l'ADN est copié avant que la cellule ne se divise.
Il existe plusieurs mécanismes de réparation de l'ADN, chacun étant spécialisé dans la correction de certains types de dommages à l'ADN. Les principaux types de réparation de l'ADN comprennent :
1. Réparation par excision de nucléotides (NER) : Ce processus répare les dommages causés aux segments individuels de la chaîne d'ADN en coupant et en éliminant les nucléotides endommagés, puis en remplaçant les nucléotides manquants par des nucléotides non endommagés.
2. Réparation de jonction d'extrémités ouverte (NHEJ) : Ce processus répare les dommages aux extrémités des brins d'ADN en joignant simplement les extrémités ensemble, souvent avec une petite perte de matériel génétique.
3. Réparation par excision de base (BER) : Ce processus répare les dommages causés à un seul nucléotide en coupant et en éliminant le nucléotide endommagé, puis en remplaçant le nucléotide manquant.
4. Réparation de recombinaison homologue (HRR) : Ce processus répare les dommages plus graves à l'ADN en utilisant une molécule d'ADN non endommagée comme modèle pour remplacer le matériel génétique manquant ou endommagé.
Les défauts dans ces systèmes de réparation peuvent entraîner des mutations et des maladies, telles que les cancers. Les chercheurs étudient actuellement comment améliorer ces systèmes de réparation pour prévenir ou traiter les maladies.
Les maladies du système nerveux sont des affections qui affectent la structure ou la fonction du système nerveux, qui est composé du cerveau, de la moelle épinière et des nerfs périphériques. Ces maladies peuvent être causées par des infections, des traumatismes, des tumeurs, des anomalies congénitales, des troubles métaboliques ou dégénératifs, ou encore des facteurs environnementaux et génétiques.
Les symptômes des maladies du système nerveux peuvent varier considérablement en fonction de la région affectée du système nerveux et de la nature de la lésion. Ils peuvent inclure des douleurs, des faiblesses musculaires, des engourdissements, des picotements, des tremblements, des convulsions, des mouvements anormaux, des problèmes de coordination, des difficultés d'élocution, des troubles cognitifs, des changements de comportement et des pertes de conscience.
Les maladies du système nerveux peuvent être classées en deux grandes catégories : les maladies du système nerveux central (qui comprennent le cerveau et la moelle épinière) et les maladies du système nerveux périphérique (qui comprennent les nerfs situés en dehors du cerveau et de la moelle épinière).
Parmi les exemples de maladies du système nerveux central, on peut citer les accidents vasculaires cérébraux, les tumeurs cérébrales, l'encéphalite, la méningite, la sclérose en plaques et la maladie d'Alzheimer.
Parmi les exemples de maladies du système nerveux périphérique, on peut citer la neuropathie diabétique, le syndrome du canal carpien, la sclérose latérale amyotrophique (SLA) et la polyneuropathie inflammatoire démyélinisante chronique (PIDC).
La checkpoint kinase 2, ou Chk2, est une protéine kinase qui joue un rôle crucial dans la régulation des points de contrôle du cycle cellulaire et de la réponse aux dommages à l'ADN. Elle est activée en réponse à des dommages à l'ADN, tels que ceux causés par les radiations ou certains agents chimothérapeutiques, et contribue à déclencher une cascade de réactions qui entraînent l'arrêt du cycle cellulaire et la réparation de l'ADN.
Lorsque Chk2 est activée, elle phosphoryle d'autres protéines, ce qui modifie leur fonction et permet de stabiliser le génome en empêchant la division cellulaire jusqu'à ce que les dommages à l'ADN soient réparés. Si les dommages sont trop graves ou ne peuvent être réparés, Chk2 peut également déclencher des processus de mort cellulaire programmée (apoptose) pour éliminer la cellule endommagée et prévenir la transformation maligne.
Des mutations dans le gène codant pour Chk2 ont été associées à un risque accru de développer certains cancers, tels que les cancers du sein et de l'ovaire. Cependant, des recherches sont en cours pour déterminer si l'inhibition ou l'activation de Chk2 pourrait être bénéfique dans le traitement de certaines maladies, y compris le cancer.
Le rayonnement ionisant est un type de radiation capable de produire ions ou paires d'ions en traversant la matière. Il possède suffisamment d'énergie pour arracher des électrons des atomes ou des molécules, ce qui les charge électriquement et les ionise.
Ce type de rayonnement comprend des radiations telles que les rayons X, les rayons gamma, les particules bêta (électrons à haute vitesse) et les particules alpha (noyaux d'hélium). L'ionisation produite par ces radiations peut endommager les cellules vivantes, altérer l'ADN et provoquer des mutations génétiques, entraînant divers effets biologiques néfastes, y compris des dommages tissulaires et des risques accrus de cancer.
Les sources de rayonnement ionisant peuvent être naturelles, comme le rayonnement cosmique et les radionucléides présents dans le sol, l'eau et l'air, ou artificielles, provenant d'activités humaines telles que la médecine nucléaire, la radiothérapie, l'imagerie médicale, l'industrie nucléaire et les centrales électriques. La protection contre le rayonnement ionisant implique généralement une réduction de l'exposition en limitant l'accès aux sources de rayonnement, en utilisant des boucliers protecteurs ou en minimisant la durée d'exposition.
La dégénérescence nerveuse est un terme général utilisé en médecine pour décrire une condition où les nerfs du corps se détériorent ou se décomposent. Cela peut se produire en raison de divers facteurs, tels que des maladies, des traumatismes, l'âge ou des habitudes malsaines.
La dégénérescence nerveuse peut affecter n'importe quel type de nerfs dans le corps, y compris les nerfs sensoriels (qui transmettent des sensations telles que la douleur, le toucher et la température), les nerfs moteurs (qui contrôlent les mouvements musculaires) et les nerfs autonomes (qui régulent les fonctions automatiques du corps telles que la fréquence cardiaque, la pression artérielle et la digestion).
Les symptômes de la dégénérescence nerveuse varient en fonction de la zone affectée et peuvent inclure des douleurs, des picotements, une faiblesse musculaire, une perte d'équilibre, une vision floue, des problèmes auditifs, des difficultés à avaler ou à parler, et une perte de contrôle de la vessie ou des intestins.
Le traitement de la dégénérescence nerveuse dépend de la cause sous-jacente. Dans certains cas, il peut être possible de ralentir ou d'arrêter la progression de la maladie grâce à des médicaments, une thérapie physique, une intervention chirurgicale ou d'autres traitements. Cependant, dans d'autres cas, la dégénérescence nerveuse peut être irréversible et entraîner des dommages permanents aux nerfs.
Une fasciculation est un type de contraction musculaire involontaire qui est généralement décrit comme une « secousse » ou une « contraction » rapide et souvent visible sous la peau. Elle se produit lorsque des faisceaux individuels de fibres musculaires se contractent de manière incontrôlable. Contrairement aux crampes musculaires, les fasciculations sont généralement indolores, bien que leur apparition fréquente ou persistante puisse être un signe de trouble neuromusculaire sous-jacent. Les causes courantes des fasciculations comprennent le stress, la fatigue, la consommation de caféine ou d'autres stimulants, et certaines maladies neurologiques telles que la sclérose latérale amyotrophique (SLA) ou la maladie de Charcot. Cependant, dans de nombreux cas, les fasciculations peuvent être bénignes et n'avoir aucune cause sous-jacente grave.
Le syndrome de Miller Fisher est un type rare de neuropathie, qui est une condition qui affecte les nerfs. Il s'agit d'une forme de triparésie, ce qui signifie qu'il implique la paralysie ou la faiblesse des trois paires de nerfs crâniens : l'oculomotrice, l'abducens et le trochléaire. Ces nerfs sont responsables du mouvement des yeux et de la coordination des mouvements oculaires avec les mouvements de la tête.
Les symptômes typiques du syndrome de Miller Fisher comprennent une paralysie des muscles oculaires, ce qui entraîne une vision double ou une difficulté à bouger les yeux, une perte d'équilibre et de coordination, ainsi qu'une absence de réflexes tendineux profonds (un signe connu sous le nom d'aréflexie). Les personnes atteintes du syndrome de Miller Fisher peuvent également présenter des difficultés à avaler et une faiblesse généralisée des muscles du visage et du corps.
Le syndrome de Miller Fisher est généralement considéré comme une forme rare de la maladie de Guillain-Barré, qui est un trouble auto-immun dans lequel le système immunitaire attaque les nerfs périphériques. Il est souvent précédé d'une infection virale ou bactérienne, telles que la grippe ou une infection à Campylobacter jejuni.
Le diagnostic du syndrome de Miller Fisher repose généralement sur l'examen clinique et les antécédents médicaux du patient, ainsi que sur des tests de laboratoire tels qu'une analyse de sang et une ponction lombaire pour examiner le liquide céphalo-rachidien. Le traitement du syndrome de Miller Fisher consiste généralement en un traitement de support et de réadaptation, ainsi que des corticostéroïdes ou des immunoglobulines intraveineuses pour aider à réduire l'inflammation et à accélérer la récupération.
En génétique, un hétérozygote est un individu qui possède deux allèles différents d'un même gène sur les deux chromosomes homologues. Cela signifie que l'individu a hérité d'un allèle particulier du gène en question de chacun de ses parents, et ces deux allèles peuvent être différents l'un de l'autre.
Dans le contexte de la génétique mendélienne classique, un hétérozygote est représenté par une notation avec une lettre majuscule suivie d'un signe plus (+) pour indiquer que cet individu est hétérozygote pour ce gène spécifique. Par exemple, dans le cas d'un gène avec deux allèles A et a, un hétérozygote serait noté Aa.
La présence d'hétérozygotie peut entraîner des phénotypes variés, en fonction du type de gène concerné et de la nature des allèles en présence. Dans certains cas, l'allèle dominant (généralement représenté par une lettre majuscule) détermine le phénotype, tandis que dans d'autres cas, les deux allèles peuvent contribuer au phénotype de manière égale ou interactive.
Il est important de noter qu'être hétérozygote pour certains gènes peut conférer des avantages ou des inconvénients en termes de santé, de résistance aux maladies et d'autres caractéristiques. Par exemple, l'hétérozygotie pour certaines mutations associées à la mucoviscidose (fibrose kystique) peut offrir une protection contre certaines bactéries nocives de l'appareil respiratoire.
La définition médicale des "Myoclonies Progessives" ou "Epilepsies Myocloniques Progressives" (EMP) est un groupe de troubles neurologiques caractérisés par la présence de myoclonies, qui sont des secousses musculaires brusques et involontaires, ainsi que d'autres types de crises épileptiques. Les myoclonies peuvent affecter n'importe quel muscle du corps, mais elles sont souvent observées dans les membres supérieurs et inférieurs.
Les EMP sont considérées comme progressives car les symptômes ont tendance à s'aggraver avec le temps. Les personnes atteintes de ces troubles peuvent présenter des difficultés croissantes pour effectuer des tâches quotidiennes, telles que marcher, parler ou manger, en raison de l'aggravation des myoclonies et d'autres symptômes.
Les EMP peuvent être causées par diverses affections sous-jacentes, notamment des maladies génétiques, des infections cérébrales, des traumatismes crâniens ou des tumeurs cérébrales. Dans certains cas, la cause peut rester inconnue même après une évaluation approfondie.
Le traitement des EMP dépend de la cause sous-jacente et peut inclure des médicaments anticonvulsivants pour contrôler les crises épileptiques, ainsi que des thérapies de réadaptation pour aider à gérer les difficultés fonctionnelles. Dans certains cas, une intervention chirurgicale peut être envisagée pour traiter les foyers épileptogènes spécifiques dans le cerveau.
La liaison génétique est un phénomène dans lequel des gènes ou des marqueurs génétiques spécifiques situés à proximité les uns des autres sur un chromosome ont tendance à être hérités ensemble au cours de la méiose, car il est moins probable qu'ils soient séparés par recombinaison génétique. Plus la distance entre deux gènes ou marqueurs est petite, plus ils sont susceptibles d'être co-transmis et donc considérés comme étant liés. La mesure de cette liaison est exprimée en unités de carte génétique, telles que les centimorgans (cM), qui représentent environ un échange réciproque par recombinaison sur 100 meioses.
Ce concept est fondamental dans la cartographie génétique et l'analyse de l'expression des gènes, ainsi que dans l'identification des mutations causales de maladies monogéniques et complexes. Il permet également d'identifier des susceptibilités génétiques à certaines conditions médicales ou traits, ce qui peut conduire à un counseling génétique plus précis et à une médecine personnalisée.
Les réflexes anormaux sont des réponses involontaires et inappropriées du corps à des stimuli normaux, dus à des perturbations du système nerveux. Ils peuvent être un signe de divers troubles neurologiques ou traumatismes. Les réflexes anormaux peuvent se manifester par des mouvements excessifs, absents ou inadéquats. Par exemple, certains réflexes présents à la naissance qui devraient disparaître avec le développement normal du cerveau, comme le réflexe de Babinski, peuvent persister en signe d'anomalie sous-jacente. D'autres réflexes anormaux peuvent apparaître à la suite d'une lésion cérébrale ou médullaire, tels que les réflexes pathologiques des membres supérieurs et inférieurs dans la paralysie cérébrale.
La paraplégie spasmodique héréditaire, également connue sous le nom de maladie de Strümpell-Lorrain ou de la maladie de Little, est une forme rare de paralysie spastique héréditaire. Il s'agit d'une maladie neurodégénérative qui affecte principalement les voies nerveuses qui contrôlent les mouvements volontaires des jambes.
Cette condition est causée par une mutation génétique dans le gène SPG4, qui code pour la protéine spastine. Cette protéine joue un rôle crucial dans le maintien de la santé des axones, les prolongements nerveux qui permettent aux neurones de communiquer entre eux. Lorsque cette protéine est défectueuse, les axones se dégénèrent progressivement, entraînant une perte de la fonction motrice et sensorielle dans les jambes.
Les symptômes de la paraplégie spasmodique héréditaire commencent généralement à apparaître pendant l'enfance ou l'adolescence, bien que dans certains cas, ils puissent ne se manifester qu'à l'âge adulte. Les premiers signes comprennent souvent une raideur musculaire accrue (spasticité), des mouvements involontaires des jambes (spasmes) et une démarche rigide ou boiteuse. Au fil du temps, ces symptômes peuvent s'aggraver, entraînant une perte de la capacité à marcher et éventuellement une paralysie complète des jambes.
Actuellement, il n'existe pas de traitement curatif pour la paraplégie spasmodique héréditaire. Le traitement est principalement axé sur la gestion des symptômes et peut inclure des médicaments pour réduire la spasticité musculaire, une thérapie physique pour maintenir la force et la flexibilité des muscles, et des dispositifs d'assistance à la mobilité tels que des fauteuils roulants.
La consanguinité, en termes médicaux, se réfère à la pratique d'avoir des relations sexuelles ou de se marier entre individus qui sont apparentés de près, ce qui peut augmenter la probabilité de naissance d'enfants avec des anomalies génétiques et certaines maladies héréditaires. Cela est dû au fait que les gènes récessifs nocifs ont une plus grande chance d'être présents dans les deux copies du gène (une copie de chaque parent) chez les enfants issus de ces unions, ce qui peut entraîner leur expression et la manifestation de problèmes de santé.
Plus le taux de consanguinité est élevé (c'est-à-dire, plus les individus sont étroitement liés), plus le risque de maladies génétiques récessives est grand chez leur progéniture. Des mariages entre cousins germains, par exemple, présentent un risque légèrement accru de certains problèmes de santé congénitaux, mais ce risque n'est généralement pas aussi élevé que dans le cas d'unions entre frères et sœurs ou parents plus proches.
Il est important de noter que la consanguinité existe depuis longtemps dans l'histoire humaine pour des raisons sociales, culturelles et géographiques. Cependant, les professionnels de la santé doivent être conscients de ce risque accru de problèmes de santé génétiques lorsqu'ils traitent des patients issus de ces unions consanguines et offrir des conseils appropriés en matière de planification familiale et de dépistage prénatal.
Les noyaux du cervelet, également connus sous le nom de noyaux profonds ou noyaux vestibulaires, sont des groupes de neurones situés dans la région centrale du cervelet. Ils jouent un rôle crucial dans l'intégration et le traitement des informations sensorielles et motrices pour coordonner les mouvements volontaires et involontaires.
Il existe quatre principaux noyaux du cervelet :
1. Noyau fastigial : situé dans la partie médiane du cervelet, il reçoit des afférences principalement du cortex cérébelleux et du tronc cérébral. Il est impliqué dans le contrôle de la posture, de l'équilibre et des mouvements oculaires.
2. Noyau globose : situé latéralement au noyau fastigial, il reçoit des afférences du cortex cérébelleux et des noyaux vestibulaires du tronc cérébral. Il contribue aux mouvements oculaires et à la coordination des membres supérieurs.
3. Noyau emboliforme : situé latéralement au noyau globose, il reçoit également des afférences du cortex cérébelleux et des noyaux vestibulaires. Il est responsable de la coordination des mouvements des membres supérieurs et inférieurs.
4. Noyau dentelé : le plus latéral et le plus volumineux des noyaux, il reçoit des afférences principalement du cortex cérébelleux et est responsable de la planification et de l'exécution des mouvements complexes, tels que ceux nécessaires à l'écriture ou à la manipulation d'objets.
Les noyaux du cervelet sont interconnectés avec d'autres structures cérébrales, telles que le thalamus et le cortex moteur, formant des boucles de rétroaction qui permettent une régulation fine et un contrôle précis des mouvements volontaires.
L'épilepsie myoclonique est un type d'épilepsie caractérisé par des spasmes musculaires brusques et involontaires appelés myoclonies. Ces spasmes peuvent affecter une seule partie du corps ou tout le corps et peuvent varier en fréquence de quelques fois par jour à plusieurs fois par semaine.
Les épilepsies myocloniques peuvent être causées par divers facteurs, y compris des anomalies génétiques, des lésions cérébrales, certaines infections ou intoxications. Dans certains cas, la cause peut rester inconnue.
Ce type d'épilepsie peut se manifester à tout âge, mais il est plus fréquent chez les enfants et les adolescents. Les épilepsies myocloniques peuvent être associées à d'autres symptômes, tels que des absences, des convulsions tonico-cloniques ou une déficience intellectuelle.
Le diagnostic d'épilepsie myoclonique repose sur l'observation des crises, la réalisation d'un électroencéphalogramme (EEG) et l'exclusion de toute autre cause possible de myoclonies. Le traitement dépend de la gravité et de la fréquence des crises, mais peut inclure des médicaments antiépileptiques, une alimentation spéciale ou, dans certains cas, une intervention chirurgicale.
Il est important de noter que les épilepsies myocloniques peuvent être bien contrôlées avec un traitement approprié, mais sans traitement, elles peuvent avoir des répercussions importantes sur la qualité de vie et le fonctionnement quotidien des personnes atteintes.
L'intelligence désigne les capacités d'une personne à apprendre, à raisonner, à résoudre des problèmes, à faire preuve de jugement et de pensée abstraite. Un handicap intellectuel, également connu sous le nom de déficience intellectuelle ou retard mental, est un trouble du développement qui affecte ces capacités intellectuelles et la capacité d'une personne à fonctionner de manière indépendante dans la vie quotidienne.
Il est généralement diagnostiqué avant l'âge de 18 ans et peut varier de léger à sévère. Les personnes atteintes de handicap intellectuel peuvent avoir des difficultés à acquérir et à appliquer de nouvelles connaissances, à communiquer efficacement, à prendre soin d'elles-mêmes, à établir des relations sociales et à faire face aux situations stressantes.
Les causes du handicap intellectuel peuvent être génétiques, environnementales ou résulter de complications pendant la grossesse ou la naissance. Il est important de noter que les personnes atteintes de handicap intellectuel ont des capacités et des besoins uniques, et qu'un diagnostic précoce et une intervention appropriée peuvent améliorer considérablement leur qualité de vie et leurs perspectives d'avenir.
L'ophtalmoplégie est un terme médical qui décrit une paralysie ou une faiblesse significative des muscles oculaires. Cela peut affecter différents muscles, entraînant une variété de symptômes. Les plus courants sont la limitation du mouvement des yeux, la déviation des yeux vers le haut, le bas, l'intérieur ou l'extérieur, et dans certains cas, la double vision.
Cette condition peut être causée par une variété de facteurs, y compris des troubles neurologiques, des infections, des traumatismes, des tumeurs ou des affections systémiques telles que le diabète et la maladie de Basedow. Le traitement dépendra de la cause sous-jacente. Dans certains cas, il peut s'agir d'une complication d'une autre condition médicale qui nécessite un traitement spécifique. Dans d'autres cas, le traitement peut viser à renforcer les muscles oculaires ou à corriger la double vision.
L'atrophie optique est un terme utilisé en ophtalmologie pour décrire la dégénérescence ou la mort des cellules nerveuses dans le nerf optique, qui relie l'œil au cerveau. Cette condition peut entraîner une perte de vision progressive ou soudaine, selon la cause sous-jacente. Les symptômes courants comprennent une réduction du champ visuel, une diminution de la acuité visuelle, des couleurs décolorées et une sensibilité accrue à la lumière.
Les causes les plus courantes d'atrophie optique comprennent les maladies neurologiques telles que la sclérose en plaques et la neuropathie optique héréditaire de Leber, ainsi que les lésions oculaires et les glaucomes non contrôlés. Dans certains cas, la cause peut être inconnue, ce qui est appelé atrophie optique idiopathique.
Le diagnostic d'atrophie optique est généralement posé par un ophtalmologiste après un examen complet de l'œil et des tests spéciaux tels qu'une visualisation du fond d'œil, une tomographie par cohérence optique (OCT) et des potentiels évoqués visuels (PEV). Le traitement dépend de la cause sous-jacente et peut inclure des médicaments, des chirurgies ou des thérapies de réadaptation visuelle.
L'hypoalbuminémie est un terme médical qui décrit une concentration anormalement faible de l'albumine sérique, une protéine produite par le foie, dans le sang. L'albumine joue un rôle crucial dans la régulation de la pression oncotique (la force osmotique due aux protéines plasmatiques) et agit comme un transporteur pour plusieurs molécules, telles que les hormones stéroïdes, les acides gras, les ions calcium et le bilirubine.
Une concentration sérique d'albumine inférieure à 3,5 g/dL est généralement considérée comme une hypoalbuminémie. Cette condition peut être liée à diverses affections sous-jacentes, y compris une mauvaise nutrition, des maladies inflammatoires chroniques, une insuffisance hépatique, une malabsorption intestinale et des maladies rénales.
L'hypoalbuminémie peut entraîner plusieurs complications, telles qu'un œdème (en raison d'une pression oncotique réduite), une susceptibilité accrue aux infections en raison de la diminution des mécanismes de défense non spécifiques et une mauvaise cicatrisation des plaies. Par conséquent, il est important d'identifier et de traiter rapidement les causes sous-jacentes de l'hypoalbuminémie pour prévenir ces complications potentielles.
Je suis désolé, mais la phrase "Maladies Expérimentales" ne correspond pas à une définition médicale spécifique. Le terme "maladie expérimentale" peut faire référence à des études ou recherches scientifiques dans lesquelles des maladies sont simulées ou créées dans un laboratoire, généralement sur des animaux de laboratoire, pour mieux comprendre la pathogenèse, le développement et les traitements possibles d'une maladie. Cependant, il ne s'agit pas d'un terme médical standardisé. Si vous cherchez des informations sur une procédure ou un concept médical spécifique, veuillez me fournir plus de détails afin que je puisse vous aider au mieux.
Les canaux potassiques Shaw, également connus sous le nom de canaux potassiques Kv4, sont un type spécifique de canaux potassiques qui jouent un rôle crucial dans la régulation de l'excitation électrique des cellules nerveuses et musculaires.
Ces canaux potassiques sont responsables du courant potassique transitoire qui se produit au début de l'activité membranaire, ce qui permet de raccourcir la durée de l'action potentiale (potentiel d'action) et de réguler la fréquence des potentiels d'action. Les canaux potassiques Shaw sont composés de plusieurs sous-unités protéiques différentes qui s'assemblent pour former un complexe fonctionnel.
Les mutations dans les gènes codant pour ces sous-unités peuvent entraîner des maladies génétiques graves, telles que le syndrome du QT long, une affection cardiaque héréditaire caractérisée par des anomalies de la repolarisation cardiaque et un risque accru d'arythmies cardiaques potentiellement mortelles. Les canaux potassiques Shaw sont donc des cibles importantes pour le développement de nouveaux traitements pharmacologiques pour les maladies cardiovasculaires et neurologiques.
Un allèle est une forme alternative d'un gène donné qui est localisé à la même position (locus) sur un chromosome homologue. Les allèles peuvent produire des protéines ou des ARNm avec des séquences différentes, entraînant ainsi des différences phénotypiques entre les individus.
Les gènes sont des segments d'ADN qui contiennent les instructions pour la production de protéines spécifiques ou pour la régulation de l'expression génique. Chaque personne hérite de deux copies de chaque gène, une copie provenant de chaque parent. Ces deux copies peuvent être identiques (homozygotes) ou différentes (hétérozygotes).
Les allèles différents peuvent entraîner des variations subtiles dans la fonction protéique, ce qui peut se traduire par des différences phénotypiques entre les individus. Certaines de ces variations peuvent être bénéfiques, neutres ou préjudiciables à la santé et à la survie d'un organisme.
Les allèles sont importants en génétique car ils permettent de comprendre comment des caractères héréditaires sont transmis d'une génération à l'autre, ainsi que les mécanismes sous-jacents aux maladies génétiques et aux traits complexes.
Les données de séquence moléculaire se réfèrent aux informations génétiques ou protéomiques qui décrivent l'ordre des unités constitutives d'une molécule biologique spécifique. Dans le contexte de la génétique, cela peut inclure les séquences d'ADN ou d'ARN, qui sont composées d'une série de nucléotides (adénine, thymine, guanine et cytosine pour l'ADN; adénine, uracile, guanine et cytosine pour l'ARN). Dans le contexte de la protéomique, cela peut inclure la séquence d'acides aminés qui composent une protéine.
Ces données sont cruciales dans divers domaines de la recherche biologique et médicale, y compris la génétique, la biologie moléculaire, la médecine personnalisée, la pharmacologie et la pathologie. Elles peuvent aider à identifier des mutations ou des variations spécifiques qui peuvent être associées à des maladies particulières, à prédire la structure et la fonction des protéines, à développer de nouveaux médicaments ciblés, et à comprendre l'évolution et la diversité biologique.
Les technologies modernes telles que le séquençage de nouvelle génération (NGS) ont rendu possible l'acquisition rapide et économique de vastes quantités de données de séquence moléculaire, ce qui a révolutionné ces domaines de recherche. Cependant, l'interprétation et l'analyse de ces données restent un défi important, nécessitant des méthodes bioinformatiques sophistiquées et une expertise spécialisée.
Les cassures double brin de l'ADN sont une forme grave de dommage à l'acide désoxyribonucléique (ADN), qui est le matériel génétique présent dans les cellules. Contrairement aux cassures simples brin, où une seule chaîne d'ADN est endommagée, les cassures double brin impliquent la rupture des deux brins d'ADN au même endroit.
Ces dommages peuvent être causés par divers facteurs, notamment l'exposition aux radiations ionisantes, certains agents chimiques et certaines enzymes présentes dans les cellules. Les cassures double brin de l'ADN peuvent entraîner des mutations génétiques, des modifications de l'expression des gènes et même la mort cellulaire si elles ne sont pas réparées correctement.
Le processus de réparation des cassures double brin de l'ADN implique plusieurs mécanismes différents, notamment la recombinaison homologue et la jonction non homologue des extrémités. Ces processus permettent de restaurer l'intégrité de l'ADN en réparant les dommages ou en éliminant les cellules endommagées si la réparation est impossible.
Les cassures double brin de l'ADN sont particulièrement préoccupantes dans les cellules souches et les cellules germinales, car elles peuvent entraîner des mutations héréditaires qui peuvent être transmises aux générations futures. Des niveaux élevés de cassures double brin de l'ADN ont été associés à un risque accru de maladies dégénératives, de cancer et d'autres affections liées au vieillissement.
Les chromosomes humains de la paire 19, également connus sous le nom de chromosomes 19, sont l'une des 23 paires de chromosomes présentes dans les cellules humaines. Chaque personne hérite d'une copie de chaque chromosome de chaque parent, ce qui signifie que nous avons deux copies du chromosome 19 en tout.
Le chromosome 19 est l'un des plus grands chromosomes humains et contient un grand nombre de gènes, estimés à environ 1 400. Il code pour une variété de protéines et de molécules impliquées dans divers processus biologiques, tels que le métabolisme, la réponse immunitaire, la fonction nerveuse et la croissance cellulaire.
Des variations dans les gènes situés sur le chromosome 19 ont été associées à un certain nombre de maladies génétiques, notamment la maladie d'Alzheimer, la thrombose veineuse profonde, la sclérose latérale amyotrophique et la surdité neurosensorielle. Les chercheurs continuent d'étudier le chromosome 19 pour comprendre son rôle dans la santé humaine et les maladies.
Friedreich3
- Syndrome de Joubert Ataxie cérébelleuse AVED ataxie avec déficit isolé en vitamine E Ataxie de Friedreich : maladie génétique chronique à transmission autosomique récessive. (wikipedia.org)
- L'ataxie de Friedreich, la plus fréquente des ataxies héréditaires, est une maladie neurodégénérative progressive incurable à ce jour. (inserm.fr)
- Une équipe Inserm mise sur la thérapie génique pour freiner la dégénérescence neuronale qui touche les personnes atteintes d' ataxie de Friedreich . (inserm.fr)
Personnes atteintes1
- Grâce aux dons, Ataxie Canada pourra soutenir d'autres personnes atteintes d'ataxie et améliorer leur bien-être, tout comme elle l'a fait pour Raphaël, qui a pu profiter du programme de soutien financier de la fondation lors de l'achat de son vélo adapté à sa condition en 2020 et de son fauteuil roulant tout-terrain hippocampe en 2021. (exp.com)
L'ataxie1
- https://lemagduchat.ouest-france.fr/dossier-818-ataxie-chat.html#:~:text=L'ataxie%20chez%20le%20chat,peut%20parfois%20même%20être%20soigné. (sos-siamois.com)
Fondation Claude St-Jean1
- Le 21 août dernier, cyclistes et marcheurs d'EXP se sont joints à l'équipe Raphman et ont relevé un défi pour amasser des fonds pour Ataxie Canada - Fondation Claude St-Jean, qui se tenait au parc Daniel-Johnson, à Granby. (exp.com)
Manifeste2
- Cette maladie se manifeste par des troubles de l'équilibre et de la coordination des mouvements volontaires (ataxie), mais aussi par une atteinte cardiaque, un diabète et, souvent, des troubles ostéo‑articulaires (scoliose, pieds creux). (inserm.fr)
- Une incoordination (ataxie) se manifeste, généralement au moment où l'enfant commence à marcher, mais cela peut n'apparaître que vers 4 ans. (msdmanuals.com)
D'une3
- Les troubles de l'équilibre sont plus évidents quand le patient a les yeux fermés dans le cas d'une ataxie labyrinthique. (wikipedia.org)
- Neurosarcoïdose Neurosyphilis : cette complication observée à la phase tertiaire de la syphilis est parfois responsable d'une atteinte de la moelle épinière qui réalise une ataxie dans le cadre d'un syndrome radiculo-cordonal postérieur appelé le Tabes dorsalis. (wikipedia.org)
- Il se peut que le chaton ou le chat ait eu un accident ou une complication quelconque et qu'en raison d'une blessure, une ataxie du chat se produise, mais il est également possible qu'il soit né avec le problème ou qu'il apparaisse après quelques semaines ou mois de vie. (planeteanimal.com)
Transmission autosomique1
- Ataxie cérébelleuse type 6 Ataxie de Charlevoix-Saguenay : maladie génétique chronique à transmission autosomique récessive. (wikipedia.org)
Maladies3
- Les ataxies spinocérébelleuses font partie des maladies génétiques neurodégénératives du cervelet et du tronc cérébral qui entrainent de nombreux troubles moteurs, et dont la forme la plus connue est la SCA3 aussi appelée maladie de Machado-Joseph. (inserm.fr)
- Dans cette étude, une équipe de l'Institut du cerveau et de la moelle épinière (Inserm/Sorbonne Université/ APHP) dirigée par Nathalie Cartier-Lacave s'est intéressée à un autre groupe de maladies présentant cette production de protéines à expansion de polyglutamines, les ataxies spinocérébelleuses, et plus spécifiquement la SCA3. (inserm.fr)
- La Filière de Santé Maladies Rares BRAIN -TEAM a proposé deux représentations du spectacle « Voyage en Ataxie » les 8 et 9 juin 2022 dernier au sein de la chapelle Saint-Louis à l'Hôpital de la Pitié Salpêtrière. (brain-team.fr)
Convulsions1
- Les signes qui justifient un retrait immédiat du terrain sont la perte de conscience réelle ou suspectée, des convulsions, une posture tonique, une ataxie, un mauvais équilibre, de la confusion, des changements de comportement et une amnésie. (medscape.com)
Font partie1
- Les ataxies font partie d'un ensemble plus grand : les troubles de l'équilibre. (wikipedia.org)
Perte1
- Dans ce nouvel article Ataxie et perte d'équilibre chez le chat - Symptômes et traitement , nous allons parler d'un problème de santé du chat domestique, plus courant qu'il ne nous semble en principe. (planeteanimal.com)
Confusion1
- Ce dysfonctionnement entraîne les premiers symptômes : KO, ataxie - le joueur titube -, confusion. (inserm.fr)
Cliniques1
- Les rares études cliniques sur le sujet ont surtout porté sur les ataxies spinocérébelleuses. (asso.fr)
L'enfant1
- Ataxie télangiectasie : cause la plus fréquente d'ataxie cérébelleuse de l'enfant dans la majorité des pays sauf au Portugal et au Japon. (wikipedia.org)
Mots1
- Cependant, une semaine après l'hospitalisation, des symptômes neurologiques prononcés sont apparus, à savoir des tremblements grossiers, un trouble de la motricité fine des deux mains, des difficultés à trouver ses mots et une ataxie. (medscape.com)
Raison1
- Pour cette raison, un trouble soit dans la moelle épinière soit dans les nerfs périphériques peut causer une ataxie. (wikipedia.org)
Musculaire1
- Désorientation, ataxie musculaire, capacité d'apprentissage affectée. (bioalaune.com)
Peut causer1
- Pour cette raison, un trouble soit dans la moelle épinière soit dans les nerfs périphériques peut causer une ataxie. (wikipedia.org)
Troubles2
- Les ataxies font partie d'un ensemble plus grand : les troubles de l'équilibre. (wikipedia.org)
- Les ataxies sont des troubles congénitaux du système nerveux central. (jack-russel.fr)
Cervelet1
- Après que la guerre Crouzon a poursuivi ses travaux sur les dystrophies héréditaires, et en particulier sur les ataxies héréditaires du cervelet, les difformités de la colonne vertébrale et les désordres rhumatismaux chroniques. (cordial.fr)
Spinale1
- L' ataxie spinale ou ataxie spinocérébelleuse (SCA) se produit de 2 à 6 mois. (jack-russel.fr)
Coordination1
- Chez le chien, le terme ataxie renvoie à une maladie neurologique qui se caractérise par une perte de coordination et de l'équilibre. (jack-russel.fr)
Maladie3
- Ataxie cérébelleuse type 6 Ataxie de Charlevoix-Saguenay : maladie génétique chronique à transmission autosomique récessive. (wikipedia.org)
- Démences Neuropathie sensitivo-motrice (Charcot-Marie-Tooth) Maladie de Hartnup Syndrome de Miller-Fisher Syndrome de Guillain-Barré Syndromes d'Ehlers-Danlos Syndrome de Wolfram Syndrome de Marinesco-Sjögren Maladie de Niemann-Pick Accident vasculaire cérébral A-bêta-lipoprotéinémie Syndrome de Shy Drager Chez les bovins, l'encéphalopathie spongiforme ("maladie de la vache folle") provoque une ataxie dans 80 % des cas. (wikipedia.org)
- Dans l' ataxie spinocérébelleuse (SCA), c'est la mutation du gène KCNJ10 qui est responsable de la maladie. (jack-russel.fr)
Niveau1
- Au niveau de l' ataxie cérébelleuse (CA), les chiots affectés ont une démarche non coordonnée, ils développent plus de difficultés à marcher vers l'avant et tombent fréquemment. (jack-russel.fr)
Moelle1
- Dans l' ataxie spinocérébelleuse (SCA) et tardive (LOA) , la moelle épinière et le cerveau sont affectés et les chiens atteints présentent des symptômes caractéristiques cliniquement très similaires. (jack-russel.fr)
Varicelle1
- Le grand frère, âgé de 6 ans maintenant, a fait à 3 ans et demi une ataxie cérébelleuse sur une varicelle. (infovac.fr)
Clinique1
- Citer cet article: Cas clinique : paresthésies, ataxie et steppage bilatéral chez un jeune patient - Medscape - 7 févr 2023. (medscape.com)
20231
- Citer cet article: Étude de cas : ataxie et hydrocéphalie chez un jeune enfant - Medscape - 28 juin 2023. (medscape.com)
Qu'à1
- Les ataxies spinocérébelleuses, qui appartiennent à la famille des ataxies héréditaires, n'entraînent des symptômes marqués qu'à l'âge adulte. (santelog.com)
Semaines2
- L' ataxie cérébelleuse (CA) se produit chez les chiots dès l'âge de 2 semaines lorsqu'ils commencent leurs premières tentatives de marche. (jack-russel.fr)
- Dans le British Medical Journal , des médecins américains rapportent le cas d'un enfant* présentant une ataxie depuis plusieurs semaines. (medscape.com)
Recherche1
- cette étude, la plus large à ce jour sur l'apparition des ataxies spinocérébelleuses, a porté sur les données de 252 adultes, tous enfants ou frères et sœurs de personnes atteintes d'ataxie spinocérébelleuse, apportées par un réseau de recherche constitué de 14 institutions scientifiques de 7 pays européens (Autriche, France, Allemagne, Hongrie, Italie, Pologne et Espagne). (santelog.com)
C'est1
- Le Défi Ataxie, c'est l'occasion de faire votre part. (fqsc.net)