Glycogénose De Type I
Glycogénose
Glycogénose De Type Iii
Glycogénose De Type Iv
Glycogénose De Type Ii
Glycogène
Glycogénose De Type Vii
Glucosephosphatase
Glycogénose De Type V
Glycogen Debranching Enzyme System
Glycogénose De Type Vi
Alpha-Glucosidase
Maladies Lysosomiales
Glycogen Synthase
Glycogénose De Type Viii
Glucan 1,4-Alpha-Glucosidase
Glucose-6-Phosphate
Antiporteurs
1,4-Alpha-Glucan Branching Enzyme
Entérocolite
Maladie De Stockage En Esters Du Cholestérol
Glycogen Phosphorylase
Aminoacidopathies Congénitales
Foie
Enzyme Replacement Therapy
Glycogénose De Type Iib
Protéines Transportant Monosaccharides
Hypoglycémie
Déficit En Fructose-1,6-Diphosphatase
Glycogen Synthase Kinase 3
Phosphorylase B
Amidon
Phosphorylases
Maladies Neurologiques De Surcharge Lysosomiale
Acide Lactique
Maladie De Surcharge En Acide Sialique
Glucose
Dependovirus
Genetic Therapy
Maladie De Wolman
Muscles Squelettiques
Polymorphisme Conformationnel Simple Brin
Mutation
Alpha-Mannosidose
Hypoxanthine
Acide Urique
Vecteur Génétique
Glycogen Synthase Kinases
Sphingolipidoses
Maladies Expérimentales
Données Séquence Moléculaire
Inosine
Exon
Lysosomes
Hypoxanthines
Souris Knockout
Maladies Des Chiens
Hétérozygote
Maladie De Gaucher
Mucopolysaccharidose De Type Vii
Séquence Nucléotidique
Généalogie
Erreurs Innées Du Métabolisme Lipidique
Mutation Ponctuelle
Mutation Faux Sens
Mucopolysaccharidose De Type I
Chiens
Séquence Des Acides Aminés
Microsomes
Lipidoses
Analyse Mutations Adn
Adenoviridae
Purines
Insuline
Fucosidose
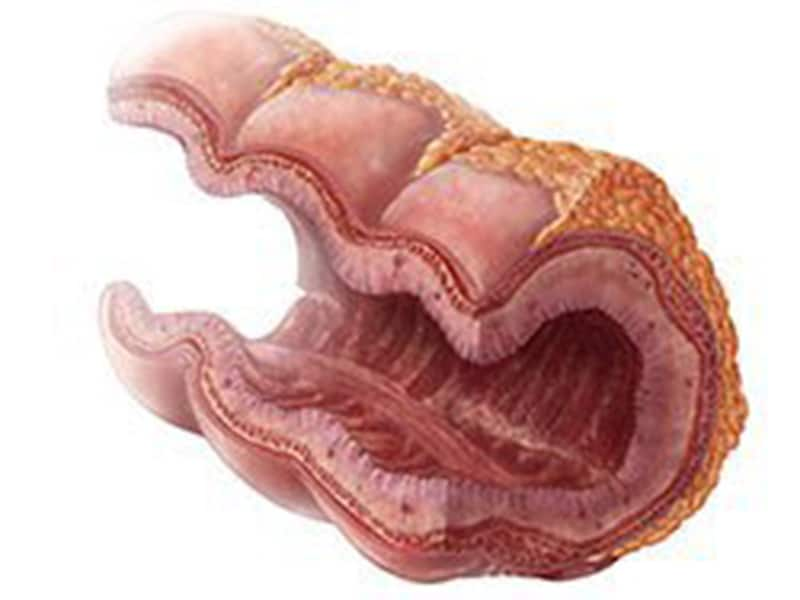
La glycogénose de type I, également connue sous le nom de maladie de von Gierke, est une maladie métabolique héréditaire rare causée par un déficit en glucose-6-phosphatase, une enzyme essentielle dans le processus de régulation de la glycémie. Cette enzyme joue un rôle clé dans la conversion du glycogène en glucose dans le foie et les reins.
En l'absence de cette enzyme, le glucose-6-phosphate s'accumule dans les cellules du foie et des reins, empêchant ainsi la libération de glucose dans la circulation sanguine. Cela entraîne une hypoglycémie à jeun sévère, une hyperlipidémie (taux élevés de graisses dans le sang), une augmentation du glycogène hépatique et une acidose métabolique due à l'accumulation d'acides organiques.
Les symptômes courants de la glycogénose de type I comprennent une croissance et un développement ralentis, une hypoglycémie, une hépatomégalie (augmentation du volume du foie), une acidose métabolique, une hyperlipidémie, une augmentation du taux d'acide urique dans le sang et une tendance aux infections.
Le diagnostic de la glycogénose de type I est généralement posé par des tests sanguins qui révèlent des niveaux anormaux de glucose, de lipides et d'acide urique. Le diagnostic définitif repose sur la confirmation de l'activité enzymatique réduite ou absente dans les échantillons de tissus hépatiques ou rénaux.
Le traitement de la glycogénose de type I implique une alimentation rigoureuse et fréquente, riche en glucides pour prévenir l'hypoglycémie, ainsi qu'un régime pauvre en graisses et en protéines pour contrôler l'hyperlipidémie et l'acidose métabolique. Des suppléments de vitamines et de minéraux peuvent également être nécessaires pour prévenir les carences nutritionnelles. Dans certains cas, des médicaments peuvent être prescrits pour traiter l'acidose métabolique et d'autres complications associées à la maladie.
La glycogénose est un terme général utilisé pour décrire un groupe d'affections métaboliques héréditaires. Ces maladies sont causées par des anomalies génétiques qui entraînent une accumulation anormale de glycogène dans les cellules du corps. Le glycogène est une forme de glucose stockée, principalement dans le foie et les muscles squelettiques.
Il existe plusieurs types de glycogénoses, chacun étant dû à une enzyme spécifique manquante ou défectueuse qui joue un rôle clé dans le métabolisme du glycogène. Selon le type, cela peut affecter divers organes et systèmes du corps, tels que le foie, les muscles squelettiques, le cœur et le cerveau.
Les symptômes de la glycogénose varient considérablement en fonction du type, allant de faibles niveaux d'énergie, une fatigue facile, des crampes musculaires à des complications plus graves telles que des lésions hépatiques, des problèmes cardiaques et même le décès. Le traitement dépend du type spécifique de glycogénose et peut inclure des changements alimentaires, des suppléments nutritionnels, des médicaments ou une transplantation d'organe dans certains cas graves.
La glycogénose de type III, également connue sous le nom de maladie de Forbes-Correll ou de déficit en glycogène débranchant, est une maladie héréditaire rare causée par une mutation du gène qui code pour l'enzyme glycogène débranchant. Cette enzyme est responsable de la dégradation du glycogène, une forme de glucose stocké dans le foie et les muscles.
Dans la glycogénose de type III, il y a un défaut de l'enzyme glycogène débranchant qui entraîne une accumulation anormale de glycogène dans les cellules du foie et des muscles. Cela peut entraîner une variété de symptômes, tels qu'une faiblesse musculaire, une hypoglycémie, une augmentation du volume du foie (hépatomégalie), une augmentation du taux de créatine kinase dans le sang et une intolérance à l'exercice.
Les symptômes peuvent varier considérablement en fonction de la sévérité de la maladie et de l'âge auquel elle se manifeste. Dans les cas graves, les bébés peuvent présenter des signes de la maladie dès la naissance, tandis que dans les cas plus légers, les symptômes peuvent ne pas apparaître avant l'enfance ou même l'âge adulte.
Le diagnostic de la glycogénose de type III est généralement posé sur la base d'un examen clinique, de tests sanguins et d'une biopsie musculaire ou hépatique pour confirmer l'accumulation anormale de glycogène. Le traitement peut inclure une alimentation riche en glucides pour prévenir les épisodes d'hypoglycémie, des suppléments de cornstarch pour maintenir un taux de sucre sanguin stable pendant la nuit et des médicaments pour traiter les complications associées à la maladie.
La glycogénose de type IV, également connue sous le nom de maladie de Andersen ou amylo-1,6-glucosidase deficiency, est une maladie héréditaire rare causée par une mutation du gène GBE1. Cette maladie affecte la capacité du corps à décomposer et à utiliser le glycogène, une forme de glucose stocké dans les muscles et le foie.
Dans la glycogénose de type IV, l'enzyme amylo-1,6-glucosidase, également appelée débranching enzyme, est déficiente ou absente. Cette enzyme joue un rôle crucial dans la décomposition du glycogène en glucose pour fournir de l'énergie aux cellules. En son absence, le glycogène s'accumule anormalement dans les organes affectés, principalement le foie et les muscles squelettiques, entraînant une dégénérescence progressive des tissus.
Les symptômes de la glycogénose de type IV peuvent varier considérablement en fonction du moment où la maladie se manifeste et de l'étendue des organes touchés. Les formes les plus graves de cette maladie peuvent provoquer une défaillance hépatique aiguë dans les premiers mois de vie, tandis que d'autres formes moins sévères peuvent ne se manifester qu'à l'âge adulte. Les symptômes courants comprennent une faiblesse musculaire progressive, une hypertrophie du foie, des vomissements, de la diarrhée, une perte de poids et un retard de croissance.
Actuellement, il n'existe aucun traitement curatif pour la glycogénose de type IV. Le traitement est principalement axé sur les symptômes et peut inclure des suppléments nutritionnels, une assistance respiratoire et une gestion des complications.
La glycogénose de type II, également connue sous le nom de maladie de Pompe, est une maladie métabolique héréditaire rare causée par une déficience en acide alpha-glucosidase (GAA), une enzyme nécessaire à la dégradation du glycogène dans les lysosomes. Le glycogène est une forme de glucose stocké dans le corps. Lorsque cette enzyme fait défaut, le glycogène s'accumule dans les lysosomes, ce qui entraîne une dégradation des cellules et des tissus, en particulier dans le cœur, le foie et les muscles squelettiques.
Les symptômes de la maladie de Pompe peuvent varier en fonction de la gravité de la déficience en GAA. Les formes les plus graves se manifestent généralement chez les nourrissons et comprennent une faiblesse musculaire grave, une hypertrophie du cœur, une insuffisance cardiaque congestive, une respiration difficile et un retard de développement. Les formes plus légères peuvent se manifester à l'âge adulte et se caractérisent par une faiblesse musculaire progressive, en particulier dans les muscles des jambes et du tronc, ainsi qu'une diminution de la fonction pulmonaire.
Le traitement de la glycogénose de type II consiste généralement en un remplacement enzymatique par une thérapie génique, qui permet de fournir une version fonctionnelle de l'enzyme GAA aux cellules affectées. Cette thérapie peut aider à ralentir la progression de la maladie et à améliorer les symptômes, mais elle ne guérit pas complètement la maladie.
Le glycogène est un polysaccharide complexe qui sert de principale forme de stockage du glucose dans les animaux, y compris les êtres humains. Il est synthétisé et stocké principalement dans le foie et les muscles squelettiques. Le glycogène hépatique peut être libéré dans la circulation sanguine pour maintenir la glycémie lorsque les réserves de glucose sont épuisées, tandis que le glycogène musculaire est utilisé localement pour fournir de l'énergie aux muscles pendant l'exercice. Le glycogène se compose de chaînes ramifiées de molécules de glucose liées entre elles par des liaisons glycosidiques. La structure ramifiée du glycogène permet une dégradation et une libération rapides de molécules de glucose lorsqu'il est nécessaire de répondre aux besoins énergétiques de l'organisme.
La glycogénose de type VII, également connue sous le nom de maladie de Tarui, est une rare maladie métabolique héréditaire causée par une mutation du gène de la phosphorylase musculaire (PYGM). Cette enzyme est responsable de la dégradation du glycogène, une forme de glucose stockée dans les muscles et le foie, en glucose-1-phosphate, qui peut être utilisé comme source d'énergie.
Dans la glycogénose de type VII, la dégradation du glycogène est altérée, entraînant une accumulation anormale de glycogène dans les muscles et un manque d'énergie disponible pour les cellules musculaires. Les symptômes de cette maladie comprennent une faiblesse musculaire sévère, une intolérance à l'exercice, des crampes musculaires, une myoglobinurie (présence de myoglobine dans les urines), une élévation des taux sanguins de créatine kinase et une augmentation du lactate sanguin après un exercice léger.
La glycogénose de type VII est généralement diagnostiquée pendant l'enfance ou l'adolescence, bien que des cas plus tardifs aient été décrits. Le diagnostic repose sur des tests génétiques et enzymatiques spécifiques. Actuellement, il n'existe pas de traitement curatif pour cette maladie, mais des mesures de soutien peuvent être mises en place pour gérer les symptômes, telles qu'une alimentation riche en glucides et une limitation de l'activité physique intense.
Je suis désolé, mais la définition d'un terme médical aussi spécifique que "glucosephosphatase" n'a pas été largement acceptée ou utilisé dans la littérature médicale. Il se peut qu'il y ait une certaine confusion avec l'enzyme « glucose-6-phosphatase », qui est un enzyme clé du métabolisme du glucose et joue un rôle crucial dans la régulation de la glycémie. Cette enzyme est responsable de la dernière étape de la gluconéogenèse et de la glycogénolyse, processus par lesquels le foie et les reins produisent du glucose à partir de précurseurs non glucidiques ou dégradent le glycogène pour fournir du glucose dans la circulation sanguine.
Cependant, comme je l'ai mentionné, "glucosephosphatase" n'est pas un terme médical reconnu et il est possible que vous ayez mal interprété ou mal orthographié le terme correct. Si vous pouviez vérifier et me fournir plus d'informations sur le contexte dans lequel ce terme a été utilisé, je serais heureux de vous fournir une réponse plus précise.
La glycogénose de type V, également connue sous le nom de maladie de McArdle, est une rare maladie métabolique héréditaire causée par une mutation du gène PYGM qui code pour l'enzyme myophosphorylase. Cette enzyme est responsable de la dégradation du glycogène, une forme de glucose stockée dans les muscles squelettiques, en glucose-1-phosphate, un précurseur de l'énergie utilisable par les cellules musculaires.
En l'absence de cette enzyme fonctionnelle, le glycogène s'accumule dans les muscles squelettiques et n'est pas décomposé en glucose-1-phosphate pour fournir de l'énergie pendant l'exercice. Cela entraîne une fatigue musculaire sévère, des crampes douloureuses et une incapacité à poursuivre l'activité physique après un effort initial. Les symptômes peuvent varier en gravité, allant de légers à graves, et peuvent inclure une intolérance à l'exercice, des douleurs musculaires, des crampes, une faiblesse musculaire et une contracture musculaire.
Le diagnostic de la glycogénose de type V est généralement posé par une biopsie musculaire qui révèle une accumulation anormale de glycogène dans les fibres musculaires squelettiques, associée à un déficit en myophosphorylase. Le traitement consiste principalement en des mesures de gestion de la fatigue et de la douleur, ainsi qu'en des conseils pour éviter les activités physiques qui déclenchent des symptômes. Les patients peuvent bénéficier d'un régime riche en glucides et d'exercices d'aérobie à faible intensité pour améliorer leur tolérance à l'exercice.
Le système d'enzymes de débranchement du glycogène est un complexe enzymatique présent dans les cellules qui joue un rôle crucial dans le métabolisme du glycogène, une forme de glucide complexe utilisée pour stocker l'énergie dans le corps. Ce système se compose de deux enzymes principales : la débranching enzyme (DBE) et la kinase dépendante de l'AMP-activée (AMPK).
La débranching enzyme, également appelée glycogène débrancheur, est responsable de l'hydrolyse des branches courtes d'unités glucose liées à la chaîne principale du glycogène. Cette enzyme clive les liaisons alpha-1,6-glucosidiques pour libérer des molécules de maltose et des chaînes plus courtes de glycogène.
La kinase dépendante de l'AMP-activée (AMPK) est une enzyme qui régule le métabolisme énergétique cellulaire en réponse aux changements de niveaux d'énergie. Elle active la débranching enzyme lorsque les niveaux d'énergie sont bas, ce qui permet au corps de libérer rapidement du glucose à partir des réserves de glycogène pour être utilisé comme source d'énergie.
Le système d'enzymes de débranchement du glycogène est important dans plusieurs tissus, y compris le foie et les muscles squelettiques, où il joue un rôle clé dans la régulation de la glycémie et de l'homéostasie énergétique. Les défauts dans ce système peuvent entraîner des maladies métaboliques telles que la glycogénose de type III, également connue sous le nom de maladie de Forbes-Correll, qui se caractérise par une accumulation anormale de glycogène dans les cellules.
La glycogénose de type VI, également connue sous le nom de maladie de Hers, est une maladie héréditaire rare causée par un déficit en activité de l'enzyme phosphorylase musculaire. Cette enzyme joue un rôle crucial dans la dégradation du glycogène, une forme de glucose stocké dans les muscles et le foie, pour produire de l'énergie lorsque le corps en a besoin.
Dans la glycogénose de type VI, le manque d'activité de l'enzyme phosphorylase musculaire entraîne une accumulation anormale de glycogène dans les muscles squelettiques et le cœur. Cela peut entraîner une faiblesse musculaire progressive, une intolérance à l'exercice, des crampes musculaires, une fatigue chronique et une myopathie (maladie musculaire).
Les symptômes de la glycogénose de type VI peuvent varier considérablement en gravité, allant de légers à sévères. Dans les cas graves, les personnes atteintes de cette maladie peuvent avoir des difficultés à marcher ou à se déplacer sans aide et peuvent nécessiter une assistance respiratoire.
Actuellement, il n'existe pas de traitement curatif pour la glycogénose de type VI. Le traitement est généralement axé sur les symptômes et peut inclure des exercices de physiothérapie, une alimentation riche en glucides et des médicaments pour soulager les crampes musculaires et la douleur. Dans certains cas, une intervention chirurgicale peut être nécessaire pour corriger les problèmes respiratoires ou cardiaques associés à la maladie.
L'alpha-glucosidase est une enzyme digestive importante qui se trouve dans l'intestin grêle des humains et d'autres mammifères. Elle joue un rôle clé dans la décomposition et l'absorption des glucides complexes, tels que l'amidon et le glycogène, en les décomposant en molécules de glucose simples qui peuvent être absorbées dans la circulation sanguine.
L'alpha-glucosidase se trouve principalement sur la surface externe des microvillosités de l'intestin grêle, où elle agit sur les molécules de glucides complexes qui ont été décomposées par d'autres enzymes digestives. En coupant les liens entre les molécules de sucre dans les glucides complexes, l'alpha-glucosidase libère des molécules de glucose simples qui peuvent être facilement absorbées dans le sang et utilisées comme source d'énergie pour le corps.
Un déficit en alpha-glucosidase peut entraîner une maladie héréditaire rare appelée la maladie de Pompe, qui est caractérisée par un stockage excessif de glycogène dans les cellules du corps. Cela peut entraîner une variété de symptômes graves, notamment une faiblesse musculaire, une hypertrophie du cœur et des difficultés respiratoires. Le traitement de la maladie de Pompe implique souvent un remplacement enzymatique, dans lequel l'alpha-glucosidase est administrée par voie intraveineuse pour aider à décomposer le glycogène accumulé dans les cellules du corps.
Les maladies lysosomales sont un groupe de plus de 50 affections héréditaires différentes causées par des mutations dans les gènes qui codent pour des protéines spécifiques, appelées enzymes lysosomiales. Ces enzymes sont responsables du processus de dégradation et de recyclage des matières cellulaires dans les lysosomes, qui sont des organites membranaires trouvés dans la plupart des cellules du corps humain.
Lorsque ces enzymes ne fonctionnent pas correctement ou manquent complètement, les molécules et les déchets cellulaires s'accumulent dans les lysosomes, entraînant une toxicité cellulaire et des dommages aux tissus et organes. Cette accumulation de matériaux peut affecter divers systèmes corporels, y compris le cerveau, la moelle osseuse, le foie, les reins, les poumons, les yeux et la peau.
Les symptômes des maladies lysosomales varient considérablement en fonction du type de maladie et de sa gravité. Ils peuvent inclure une croissance et un développement anormaux, des anomalies squelettiques, des problèmes neurologiques, des dommages aux organes internes, des problèmes sensoriels tels que la surdité ou la cécité, et une diminution de l'espérance de vie.
Les maladies lysosomales sont généralement héritées d'une manière autosomique récessive, ce qui signifie qu'un individu doit hériter de deux copies du gène muté (une de chaque parent) pour développer la maladie. Cependant, certaines formes de ces maladies peuvent être héritées d'une manière liée à l'X, ce qui signifie qu'un seul gène muté sur le chromosome X peut entraîner la maladie.
Actuellement, il n'existe pas de traitement curatif pour les maladies lysosomales, mais des thérapies enzymatiques substitutives et d'autres approches thérapeutiques peuvent aider à gérer les symptômes et à améliorer la qualité de vie des personnes atteintes. La recherche se poursuit pour développer de nouveaux traitements et thérapies pour ces maladies rares mais graves.
Le glycogène hépatique se réfère à la forme stockée des glucides dans le foie. Le foie joue un rôle crucial dans la régulation des niveaux de sucre dans le sang en convertissant l'excès de glucose sanguin en glycogène, qui est ensuite stocké dans les cellules hépatiques. Lorsque les niveaux de sucre dans le sang sont bas, comme pendant le jeûne nocturne ou entre les repas, le foie peut décomposer et libérer le glycogène sous forme de glucose dans la circulation sanguine pour maintenir une glycémie normale. Le glycogène hépatique est essentiel pour assurer un approvisionnement constant en énergie aux cellules du corps et prévenir l'hypoglycémie.
La glycogène synthase est un enzyme clé dans le métabolisme des glucides, plus spécifiquement dans la biosynthèse du glycogène. Il catalyse l'addition de molécules de glucose à une chaîne de polymères de glycogène, un processus connu sous le nom de glycogénogenèse. Cette enzyme est régulée de manière complexe, avec une activation accrue lorsque les niveaux d'insuline sont élevés et des niveaux réduits de glucagon, ce qui permet au corps de stocker efficacement le glucose excédentaire sous forme de glycogène dans le foie et les muscles squelettiques lorsque les apports en glucides sont élevés, puis de décomposer et de libérer du glucose à partir du glycogène pendant les périodes de jeûne ou d'activité physique accrue. La glycogène synthase est souvent une cible thérapeutique dans le traitement des troubles métaboliques tels que le diabète de type 2.
La glycogénose de type VIII, également connue sous le nom de déficit en phosphorylase kinase, est une maladie métabolique héréditaire rare. Cette condition affecte la capacité des cellules à décomposer le glycogène, une forme de glucose stockée dans le foie et les muscles, en glucose libre qui peut être utilisé comme source d'énergie.
Le déficit en phosphorylase kinase est causé par des mutations dans les gènes qui codent pour les sous-unités de la phosphorylase kinase, une enzyme clé dans le processus de décomposition du glycogène. Ces mutations entraînent une réduction de l'activité de la phosphorylase kinase, ce qui perturbe la capacité des cellules à convertir le glycogène en glucose-1-phosphate, un précurseur du glucose.
Les symptômes de la glycogénose de type VIII peuvent varier considérablement en fonction de la gravité de la maladie et de la partie du corps touchée. Les formes les plus graves affectent les muscles squelettiques et le foie, entraînant une faiblesse musculaire sévère, une hypotonie, une augmentation de la taille du foie (hépatomégalie), une intolérance au jeûne et une acidose métabolique. Les formes moins graves peuvent ne présenter que des symptômes légers ou même être asymptomatiques.
Le diagnostic de la glycogénose de type VIII est généralement posé sur la base d'un examen clinique, d'une analyse biochimique et d'un test génétique. Le traitement dépend de la gravité de la maladie et peut inclure une alimentation riche en glucides, des suppléments nutritionnels, des médicaments pour contrôler l'acidose métabolique et une kinésithérapie pour aider à renforcer les muscles. Dans les cas graves, une transplantation hépatique peut être envisagée.
Je suis désolé, mais la requête est un peu imprécise. Le terme "glucan 1,4-alpha-glucosidase" peut se référer à une enzyme spécifique ou à une activité enzymatique particulière. Je vous fournirai donc les deux définitions possibles :
1) En tant qu'enzyme : La glucan 1,4-alpha-glucosidase, également connue sous le nom d'amyloglucosidase ou de maltase-glucoamylase, est une enzyme qui dégrade les polysaccharides à base d'amidon, tels que le glycogène et l'amidon, en glucose. Cette enzyme clive spécifiquement les liaisons α(1→4) et α(1→6) dans ces polysaccharides complexes. L'activité de cette enzyme est importante dans la digestion des glucides chez les humains et d'autres organismes.
2) En tant qu'activité enzymatique : L'activité de la glucan 1,4-alpha-glucosidase fait référence à la capacité d'une enzyme à cliver les liaisons α(1→4) et α(1→6) dans des polysaccharides tels que le glycogène et l'amidon, libérant du glucose monomère. Cette activité est couramment détectée et mesurée en laboratoire pour étudier les propriétés de diverses enzymes et leurs applications dans des domaines tels que la biochimie, la biotechnologie et l'industrie alimentaire.
Le glucose-6-phosphate est un composé organique qui joue un rôle crucial dans le métabolisme du glucose dans les cellules vivantes. Il s'agit d'une molécule de glucose liée à une molécule de phosphate au carbone 6, ce qui permet de la maintenir active et disponible pour des processus tels que la glycolyse et la pentose phosphate pathway (PPP).
Dans le cadre du métabolisme du glucose, l'enzyme hexokinase catalyse la réaction de conversion du glucose en glucose-6-phosphate lors de son entrée dans la cellule. Cette étape est essentielle pour réguler et maintenir les niveaux de glucose intracellulaire, car le glucose libre ne peut pas traverser facilement la membrane cellulaire.
Le glucose-6-phosphate sert également d'intermédiaire dans la biosynthèse du glycogène et des lipides, ce qui en fait un acteur clé dans le stockage de l'énergie métabolique. En outre, il est impliqué dans les mécanismes de défense contre le stress oxydatif grâce à la PPP, où il est converti en pentoses et NADPH pour soutenir la synthèse des antioxydants et d'autres processus anaboliques.
En cas de dysfonctionnement du métabolisme du glucose-6-phosphate, divers troubles peuvent survenir, tels que des désordres glycogénolytiques, des défauts dans la biosynthèse des lipides et une susceptibilité accrue à l'oxydation. Par conséquent, il est essentiel de comprendre le rôle du glucose-6-phosphate dans les processus métaboliques pour diagnostiquer et traiter ces affections.
Les antiporteurs sont un type spécifique de protéines membranaires qui facilitent le transport de différentes molécules à travers la membrane cellulaire. Contrairement aux symporteurs, qui co-transportent des molécules dans la même direction, les antiporteurs échangent des molécules dans des directions opposées.
Ce processus est appelé l'antiport et permet de maintenir l'homéostasie cellulaire en régulant les niveaux de différentes molécules à l'intérieur et à l'extérieur de la cellule. Les antiporteurs peuvent être spécifiques au transport de différents ions, nutriments ou métabolites, selon le type de cellule et sa fonction.
Les antiporteurs jouent un rôle important dans de nombreux processus physiologiques, tels que la régulation du pH intracellulaire, la gestion des niveaux d'énergie et la communication intercellulaire. Cependant, ils peuvent également être impliqués dans certaines maladies, telles que les troubles neurologiques et cardiovasculaires, lorsqu'ils sont altérés ou fonctionnent de manière anormale.
Un adénome hépatocellulaire, également connu sous le nom d'hépatocellulaire adénome ou d'adénome du foie, est un type rare et généralement bénin de tumeur du foie. Il se développe à partir des cellules hépatiques (hépatocytes) qui constituent la majeure partie du foie.
Habituellement, les adénomes hépatocellulaires surviennent chez les femmes d'âge moyen, en particulier celles qui utilisent des contraceptifs oraux contenant des œstrogènes à fortes doses pendant une longue période. D'autres facteurs de risque peuvent inclure l'obésité, la maladie métabolique hépatique et certaines affections génétiques rares.
La plupart des adénomes hépatocellulaires ne présentent aucun symptôme et sont découverts de manière fortuite lors d'examens d'imagerie du foie effectués pour d'autres raisons. Cependant, certains patients peuvent présenter des douleurs abdominales, une sensation de plénitude ou une masse palpable dans l'abdomen supérieur. Les complications graves mais rares associées aux adénomes hépatocellulaires comprennent la rupture et les saignements intra-abdominaux, qui peuvent mettre la vie en danger.
Le traitement de choix pour l'adénome hépatocellulaire est généralement la chirurgie visant à retirer complètement la tumeur. Dans certains cas, une transplantation hépatique peut être recommandée, en particulier pour les patients atteints de tumeurs multiples ou de tumeurs volumineuses qui ne peuvent pas être complètement retirées par chirurgie.
Il est important de noter que bien que la plupart des adénomes hépatocellulaires soient bénins, il existe un risque qu'ils deviennent cancéreux au fil du temps. Par conséquent, les patients atteints d'adénome hépatocellulaire doivent faire l'objet d'un suivi régulier par imagerie pour détecter toute récidive ou progression de la maladie.
L'amylosectine est un type de glucide complexe (polysaccharide) qui est composé principalement de molécules de sucre appelées molécules d'amylopectine. L'amylopectine est une chaîne ramifiée d'unités de glucose et est l'un des deux principaux composants de l'amidon, avec l'amylose.
L'amylosectine se distingue de l'amylose par sa structure plus ramifiée, ce qui lui permet d'être décomposé et absorbé plus rapidement dans le corps que l'amylose. L'amylosectine est une source importante d'énergie pour l'organisme et se trouve en grande quantité dans les aliments riches en amidon, tels que les céréales, les pommes de terre et les légumes.
Il est important de noter qu'il n'existe pas de condition médicale appelée "amylopectinose". Cependant, il existe une maladie rare mais grave appelée amylose, qui est causée par l'accumulation anormale d'une protéine appelée fibrilles amyloïdes dans les tissus et les organes du corps. Cette condition n'est pas liée à la consommation ou à l'accumulation d'amylopectine dans le corps.
La 1,4-alpha-glucane branching enzyme est une enzyme qui joue un rôle clé dans la biosynthèse de l'amidon. Elle catalyse le transfert d'une chaîne de glucose liée par une liaison alpha 1,4 à une autre chaîne de glucose, créant ainsi une nouvelle liaison alpha 1,6 et formant une branche dans la molécule d'amidon. Cette enzyme est importante pour la régulation de la structure et des propriétés fonctionnelles de l'amidon, qui est un polysaccharide important stocké dans les plantes comme source d'énergie. Des mutations dans le gène codant pour cette enzyme peuvent entraîner des troubles du métabolisme de l'amidon chez l'homme.
L'entérocolite est un terme médical qui décrit une inflammation affectant à la fois l'intestin grêle (petit intestin) et le côlon (gros intestin). Cette condition peut entraîner une variété de symptômes, tels que des douleurs abdominales, de la diarrhée, du sang dans les selles, de la fièvre, des nausées et des vomissements.
Il existe plusieurs types d'entérocolite, dont l'entérocolite aiguë, qui peut être causée par une infection bactérienne ou virale, ou par une réaction immunitaire anormale chez certaines personnes atteintes de maladies inflammatoires de l'intestin (MII) telles que la maladie de Crohn et la colite ulcéreuse.
L'entérocolite chronique, quant à elle, peut être associée à des affections sous-jacentes telles que la maladie cœliaque, la maladie de Crohn ou le syndrome du côlon irritable. Dans certains cas, l'entérocolite peut également être causée par une intervention chirurgicale antérieure ou par un traumatisme abdominal.
Le traitement de l'entérocolite dépend de la cause sous-jacente et peut inclure des médicaments pour contrôler l'inflammation, des antibiotiques pour combattre une infection bactérienne, une thérapie nutritionnelle ou, dans certains cas graves, une intervention chirurgicale.
La maladie de stockage en esters du cholestérol, également connue sous le nom de maladie de Wolman ou de maladie de Wolman-Castleman, est une maladie héréditaire rare et grave qui affecte le métabolisme du cholestérol. Cette condition est causée par des mutations dans le gène LIPA, ce qui entraîne une déficience en lipase acide lysosomale, une enzyme responsable de la dégradation des esters de cholestérol et des triglycérides dans les cellules.
En raison du manque d'activité de cette enzyme, les esters de cholestérol s'accumulent dans divers organes et tissus, y compris le foie, la rate, les ganglions lymphatiques, les intestins et le cerveau. Cette accumulation entraîne une inflammation, une dégénération des tissus et un dysfonctionnement progressif des organes affectés.
Les symptômes de la maladie de stockage en esters du cholestérol apparaissent généralement dans les premiers mois de vie et peuvent inclure une augmentation du volume abdominal due à l'agrandissement du foie et de la rate, des vomissements persistants, une diarrhée sévère, une perte de poids, une jaunisse, une anémie et un retard de croissance. Les enfants atteints de cette maladie peuvent également présenter des troubles neurologiques tels que des convulsions, une microcéphalie (petite taille de la tête) et un développement intellectuel et moteur anormal.
Actuellement, il n'existe aucun traitement curatif pour cette maladie. Le traitement est principalement axé sur les symptômes et vise à améliorer la qualité de vie des patients. Les options thérapeutiques peuvent inclure une alimentation spécialisée, des suppléments nutritionnels, des médicaments pour contrôler l'inflammation et la douleur, ainsi qu'un soutien psychologique et social pour les familles concernées.
L'hépatomégalie est un terme médical qui se réfère à une augmentation anormale de la taille du foie. Le foie est situé dans la partie supérieure droite de l'abdomen et joue un rôle crucial dans de nombreuses fonctions corporelles, y compris la détoxification, le métabolisme des graisses et la production de protéines.
L'hépatomégalie peut être causée par une variété de facteurs, tels que les maladies hépatiques inflammatoires (comme l'hépatite), l'infiltration du foie par des cellules anormales (comme dans le cancer du foie), la congestion veineuse (par exemple, insuffisance cardiaque congestive), l'accumulation de graisse dans le foie (comme dans la stéatose hépatique) et d'autres affections.
Les symptômes associés à l'hépatomégalie peuvent inclure une douleur ou une gêne abdominale, une perte d'appétit, une sensation de plénitude après avoir mangé, une fatigue, une jaunisse (jaunissement de la peau et des yeux) et d'autres signes de maladie hépatique. Le diagnostic d'hépatomégalie est généralement posé par un examen physique, bien que des tests d'imagerie tels qu'une échographie ou une tomodensitométrie puissent être utilisés pour confirmer la taille du foie et exclure d'autres causes de douleur abdominale.
Le traitement de l'hépatomégalie dépendra de la cause sous-jacente et peut inclure des médicaments, une intervention chirurgicale, une modification du mode de vie ou une combinaison de ces options. Il est important de consulter un professionnel de la santé si vous ressentez des symptômes d'hépatomégalie ou si vous avez des préoccupations concernant votre foie ou votre santé globale.
La glycogène phosphorylase est un enzyme clé dans la régulation du métabolisme du glycogène, qui est la forme de stockage principale des glucides dans les animaux. Cet enzyme catalyse la phosphorolyse du glycogène, un processus qui décompose le glycogène en molécules de glucose-1-phosphate, qui peuvent ensuite être converties en glucose et utilisées comme source d'énergie pour les cellules.
La glycogène phosphorylase existe sous deux formes : la forme a, qui est active en présence de calcium et est régulée par des hormones telles que l'adrénaline et l'insuline, et la forme b, qui est moins active et prédomine dans les tissus au repos.
La dérégulation de l'activité de la glycogène phosphorylase a été associée à plusieurs maladies métaboliques, telles que le diabète et les maladies du foie.
Les aminoacidopathies congénitales sont un groupe d'affections héréditaires rares caractérisées par une accumulation toxique de certaines acides aminés dans le corps en raison d'un déficit enzymatique. Les acides aminés sont les éléments constitutifs des protéines et sont normalement métabolisés dans le foie. Cependant, lorsqu'une personne hérite d'une copie altérée de l'un des gènes responsables du métabolisme des acides aminés, cela peut entraîner une accumulation toxique de ces composés dans le corps.
Les symptômes et la gravité de ces affections varient considérablement en fonction du type d'aminoacidopathie congénitale et de l'importance du déficit enzymatique. Les symptômes peuvent apparaître à tout moment de la vie, bien que dans de nombreux cas, ils soient présents dès la naissance ou se développent au cours des premiers mois de vie.
Les exemples courants d'aminoacidopathies congénitales comprennent :
1. Phénylcétonurie (PKU) - déficit en phénylalanine hydroxylase, entraînant une accumulation de phénylalanine dans le sang et le cerveau. Les symptômes peuvent inclure des retards de développement, des convulsions, une microcéphalie, des problèmes de peau et de cheveux, ainsi que des troubles comportementaux et mentaux.
2. Tyrosinémie de type I - déficit en fumarylacétoacétase, entraînant une accumulation de tyrosine et de ses métabolites toxiques dans le foie, les reins et d'autres organes. Les symptômes peuvent inclure des vomissements, une jaunisse, une odeur d'urine anormale, une hypertension portale, une insuffisance hépatique et rénale, ainsi qu'une augmentation du risque de cancer du foie.
3. Homocystinurie - déficit en cystathionine bêta-synthase, entraînant une accumulation d'homocystéine dans le sang et les tissus. Les symptômes peuvent inclure des malformations vasculaires, des troubles de la vision, des problèmes osseux, des retards de développement et des convulsions.
4. Acidurie isovalérique - déficit en acide isovalérique déshydrogénase, entraînant une accumulation d'acide isovalérique dans le sang et l'urine. Les symptômes peuvent inclure des vomissements, une acidose métabolique, une odeur de sueur et d'urine anormale, ainsi que des problèmes neurologiques tels que des convulsions et des retards de développement.
Le traitement des aminoacidopathies congénitales implique généralement un régime alimentaire restrictif et/ou un apport supplémentaire de nutriments spécifiques, ainsi qu'une surveillance médicale étroite pour prévenir les complications. Dans certains cas, une greffe de foie peut être recommandée pour traiter l'insuffisance hépatique associée à certaines formes d'aminoacidopathies congénitales.
Le foie est un organe interne vital situé dans la cavité abdominale, plus précisément dans le quadrant supérieur droit de l'abdomen, juste sous le diaphragme. Il joue un rôle essentiel dans plusieurs fonctions physiologiques cruciales pour le maintien de la vie et de la santé.
Dans une définition médicale complète, le foie est décrit comme étant le plus grand organe interne du corps humain, pesant environ 1,5 kilogramme chez l'adulte moyen. Il a une forme et une taille approximativement triangulaires, avec cinq faces (diaphragmatique, viscérale, sternale, costale et inférieure) et deux bords (droits et gauches).
Le foie est responsable de la détoxification du sang en éliminant les substances nocives, des médicaments et des toxines. Il participe également au métabolisme des protéines, des glucides et des lipides, en régulant le taux de sucre dans le sang et en synthétisant des protéines essentielles telles que l'albumine sérique et les facteurs de coagulation sanguine.
De plus, le foie stocke les nutriments et les vitamines (comme la vitamine A, D, E et K) et régule leur distribution dans l'organisme en fonction des besoins. Il joue également un rôle important dans la digestion en produisant la bile, une substance fluide verte qui aide à décomposer les graisses alimentaires dans l'intestin grêle.
Le foie est doté d'une capacité remarquable de régénération et peut reconstituer jusqu'à 75 % de son poids initial en seulement quelques semaines, même après une résection chirurgicale importante ou une lésion hépatique. Cependant, certaines maladies du foie peuvent entraîner des dommages irréversibles et compromettre sa fonctionnalité, ce qui peut mettre en danger la vie de la personne atteinte.
L'Thérapie de Remplacement Enzymatique (TRE) est une forme de traitement médical spécifiquement utilisée pour remédier aux déficiences enzymatiques congénitales. Dans cette thérapie, la ou les enzymes manquantes ou insuffisamment produites par l'organisme sont fournies de l'extérieur, sous forme de suppléments enzymatiques. Ces enzymes exogènes sont généralement administrées par voie intraveineuse, mais elles peuvent également être administrées par d'autres voies, telles que la voie orale ou inhalée, selon le type d'enzyme et la maladie concernés.
La TRE a pour objectif de compenser le déficit enzymatique, de rétablir l'équilibre métabolique et de prévenir ou ralentir la progression des dommages tissulaires associés à la maladie sous-jacente. Elle est principalement utilisée pour traiter un certain nombre de troubles héréditaires rares, tels que la mucopolysaccharidose (MPS), la gangliosidose à GM1, la déficience en α-galactosidase A (maladie de Fabry) et la déficience en glucocérébrosidase (maladie de Gaucher).
Il est important de noter que la TRE ne guérit pas ces maladies, mais elle peut considérablement améliorer la qualité de vie des patients, ralentir la progression de la maladie et potentialement prolonger l'espérance de vie. Les effets secondaires de la TRE dépendent du type d'enzyme utilisé et peuvent inclure des réactions allergiques, des rougeurs ou des douleurs au site d'injection, ainsi que des problèmes immunitaires à long terme dans de rares cas.
La glycogénose de type IIb, également connue sous le nom de maladie de Pompe, est une maladie métabolique héréditaire due à un déficit en activité de l'acide alpha-1,4-glucosidase (également appelée maltase acide), une enzyme nécessaire à la dégradation du glycogène dans les lysosomes. Le glycogène est une forme de glucose stocké dans le corps.
Ce déficit en activité enzymatique entraîne une accumulation de glycogène dans les lysosomes des cellules, ce qui peut affecter plusieurs organes et systèmes, mais principalement le cœur et les muscles squelettiques. Les symptômes peuvent varier en fonction de la gravité de la maladie et de l'âge d'apparition, mais ils comprennent généralement une faiblesse musculaire progressive, une hypertrophie cardiaque, une insuffisance respiratoire et une atteinte hépatique.
Il existe deux formes cliniques de la maladie de Pompe : la forme infantile précoce et la forme tardive de l'enfant ou de l'adulte. La forme infantile précoce est la plus grave et peut entraîner une mort prématurée en raison d'une insuffisance cardiaque congestive sévère. La forme tardive de l'enfant ou de l'adulte se manifeste généralement par une faiblesse musculaire progressive, une atteinte respiratoire et une hypertrophie cardiaque modérée.
Le traitement de la maladie de Pompe repose sur le remplacement enzymatique par l'administration de doses régulières d'alpha-glucosidase recombinante, qui peut aider à ralentir ou à arrêter la progression de la maladie et à améliorer les symptômes.
Les protéines transportant monosaccharides, également connues sous le nom de transporteurs de glucose ou de sucre, sont un type spécifique de protéines membranaires qui facilitent le mouvement des monosaccharides (des sucres simples) à travers les membranes cellulaires.
Les monosaccharides, tels que le glucose, le fructose et le galactose, sont essentiels pour la production d'énergie dans les cellules. Cependant, en raison de leur structure moléculaire, ils ne peuvent pas traverser librement la membrane cellulaire. Par conséquent, les protéines transportant des monosaccharides jouent un rôle crucial dans le processus d'absorption et de transport des sucres simples dans et hors des cellules.
Ces protéines fonctionnent comme des canaux ou des pompes qui permettent aux molécules de monosaccharides de se déplacer passivement ou activement à travers la membrane cellulaire, en fonction du gradient de concentration des sucres. Les transporteurs de glucose peuvent être classés en deux catégories principales : les transporteurs sodium-dépendants (SGLT) et les transporteurs sodium-indépendants (GLUT).
Les transporteurs sodium-dépendants nécessitent l'utilisation d'un gradient de sodium pour faciliter le mouvement des molécules de glucose à travers la membrane cellulaire. Ce type de transporteur est principalement responsable de l'absorption du glucose dans l'intestin grêle et des reins.
Les transporteurs sodium-indépendants, quant à eux, facilitent le mouvement du glucose en fonction du gradient de concentration, sans nécessiter de gradient de sodium. Ce type de transporteur est largement répandu dans différents tissus corporels, tels que le cerveau, les muscles squelettiques et le foie.
Dans l'ensemble, les transporteurs de glucose jouent un rôle crucial dans la régulation de la glycémie et des processus métaboliques associés au glucose dans l'organisme.
L'hypoglycémie est une condition médicale où le taux de glucose sanguin (glycémie) chute à un niveau dangereusement bas, généralement inférieur à 70 mg/dL. Elle peut survenir chez les personnes atteintes de diabète lorsqu'elles prennent trop de médicaments pour abaisser leur glycémie ou sautent des repas. Chez les personnes qui ne sont pas atteintes de diabète, l'hypoglycémie peut être causée par certaines maladies du foie, des reins ou des hormones, ainsi que par la consommation d'alcool à jeun ou l'exercice physique intense sans apport adéquat de nourriture.
Les symptômes de l'hypoglycémie peuvent inclure :
- Transpiration excessive
- Palpitations cardiaques
- Tremblements ou secousses musculaires
- Faim intense
- Vertiges ou étourdissements
- Vision floue
- Difficulté à penser clairement ou confusion
- Agitation ou irritabilité
- Somnolence ou fatigue
- Maux de tête
- Nausées ou vomissements
Dans les cas graves, l'hypoglycémie peut entraîner une perte de conscience, des convulsions et même la mort si elle n'est pas traitée rapidement. Le traitement consiste généralement à consommer rapidement des aliments ou des boissons riches en glucides pour augmenter le taux de sucre dans le sang, comme du jus de fruits, du soda sucré, des bonbons ou des biscuits Graham. Si la personne est inconsciente, elle peut nécessiter une injection d'une solution de glucose par voie intraveineuse ou une injection de glucagon pour réduire rapidement la sévérité de l'hypoglycémie.
Le déficit en fructose-1,6-diphosphatase (DF-1,6-DP) est un trouble métabolique héréditaire rare caractérisé par une incapacité de l'enzyme fructose-1,6-diphosphatase à fonctionner correctement. Cette enzyme joue un rôle crucial dans la décomposition du fructose et dans le processus de gluconéogenèse (la production de glucose à partir de précurseurs non glucidiques).
Lorsque cette enzyme ne fonctionne pas correctement, le métabolisme du fructose est altéré, entraînant une accumulation de fructose-1-phosphate dans l'organisme. Cela peut conduire à une acidose métabolique, une hypoglycémie, une hyperlactacidémie et d'autres complications métaboliques graves.
Les symptômes du déficit en fructose-1,6-diphosphatase peuvent inclure des vomissements, une léthargie, une hypotonie, une acidose métabolique, une hypoglycémie sévère et, dans les cas graves, un coma ou même le décès. Ce trouble est généralement diagnostiqué pendant la petite enfance et peut être déclenché par des périodes de jeûne ou des infections.
Le traitement du déficit en fructose-1,6-diphosphatase implique généralement un régime strict d'évitement du fructose, du sorbitol et du sucre de table (saccharose), ainsi qu'une gestion attentive des épisodes métaboliques aigus. Dans certains cas, des suppléments de glucose peuvent être nécessaires pour prévenir l'hypoglycémie.
La glycogène synthase kinase 3 (GSK-3) est une protéine kinase largement distribuée dans les tissus animaux et jouant un rôle crucial dans la régulation de divers processus cellulaires, tels que la glycogénogenèse, la glycolyse, la cytosquelette, la signalisation du récepteur, l'apoptose et la transcription. Il existe deux isoformes de GSK-3, alpha et beta, qui sont codées par des gènes différents mais partagent une homologie structurelle élevée.
GSK-3 est principalement connue pour sa fonction dans la phosphorylation et l'inactivation de la glycogène synthase, une enzyme clé dans le processus de glycogénogenèse. En plus de cela, il régule également d'autres voies métaboliques, telles que la dégradation des protéines et l'homéostasie du glucose.
L'activité de GSK-3 est régulée par plusieurs mécanismes, notamment la phosphorylation réversible de résidus spécifiques dans le domaine catalytique de l'enzyme. La phosphorylation inhibitrice de la sérine 21 dans GSK-3α et de la sérine 9 dans GSK-3β entraîne une diminution de son activité, tandis que la déphosphorylation de ces résidus conduit à une activation de l'enzyme.
GSK-3 est également impliquée dans plusieurs processus pathologiques, tels que la maladie d'Alzheimer, le diabète de type 2 et certains cancers. Par conséquent, il représente une cible thérapeutique potentielle pour le développement de médicaments contre ces maladies.
La phosphorylase b est une enzyme glycogénolytique qui joue un rôle clé dans la dégradation du glycogène, une molécule de réserve d'énergie stockée principalement dans le foie et les muscles squelettiques. Cette enzyme est responsable de la libération des molécules de glucose à partir du glycogène en catalysant la réaction de phosphorolyse, où un résidu de phosphate est transféré d'un groupe phosphate activé (phosphate de pyridoxal) sur l'enzyme à un résidu glucosidique du glycogène.
La phosphorylase b est une forme inactive de l'enzyme qui peut être activée par la phosphorylation d'un résidu de sérine spécifique par une kinase, telle que la protéine-kinase A (PKA). Lorsque l'enzyme est phosphorylée et convertie en sa forme active, la phosphorylase a, elle peut participer au métabolisme énergétique en décomposant le glycogène pour produire du glucose-1-phosphate, qui peut être ultérieurement transformé en glucose-6-phosphate et utilisé dans la glycolyse pour générer de l'ATP.
La régulation de l'activité de la phosphorylase b est essentielle pour maintenir l'homéostasie du métabolisme énergétique, en particulier pendant l'exercice et le jeûne, lorsque les besoins en énergie sont accrus.
L'amidon est un polysaccharide complexe, composé de chaînes ramifiées d'unités de glucose, que l'on trouve principalement dans les grains et les tubercules des plantes. Il sert de réserve d'énergie pour la plante et est souvent utilisé comme agent de liaison ou d'épaississement dans l'industrie alimentaire.
Dans le contexte médical, l'amidon peut être utilisé comme source de glucides à libération lente dans les régimes alimentaires pour aider à contrôler la glycémie. Il est également utilisé dans certains médicaments oraux pour prolonger la durée d'action du médicament dans le corps en ralentissant sa libération.
Cependant, il est important de noter que l'amidon peut être mal toléré chez certaines personnes atteintes de troubles digestifs tels que le syndrome de l'intestin irritable ou l'intolérance au fructose, car leur système digestif a du mal à décomposer et à absorber ce glucide complexe.
Les phosphorylases sont des enzymes qui catalysent la réaction de phosphorolyse, c'est-à-dire la réaction dans laquelle un groupe moléculaire de phosphate est transféré d'une molécule à une autre. Plus spécifiquement, les phosphorylases décomposent des composés tels que le glycogène ou le poly saccharides en retirant des unités monosaccharides et en les transformant simultanément en dérivés phosphorylés.
Dans le cas du glycogène, qui est une chaîne de glucose, la phosphorylase retire les unités de glucose de l'extrémité non réductrice de la chaîne et les transforme en dérivés glucose-1-phosphate. Cette réaction libère également un groupe moléculaire d'inorganique phosphate.
Les phosphorylases jouent un rôle important dans le métabolisme des glucides, en particulier pendant l'exercice physique et le jeûne, lorsque les cellules ont besoin de libérer rapidement de l'énergie à partir du glycogène stocké. Il existe plusieurs types de phosphorylases, y compris la glycogène phosphorylase qui se trouve dans les muscles et le foie.
Les maladies neurologiques de surcharge lysosomiale (NLLD) représentent un groupe de troubles héréditaires rares causés par des mutations génétiques qui entraînent une accumulation toxique de divers substrats dans les lysosomes. Les lysosomes sont des organites cellulaires responsables de la dégradation et du recyclage des matières intracellulaires. Lorsque ces enzymes lysosomales ne fonctionnent pas correctement, les substrats ne peuvent pas être correctement décomposés et s'accumulent dans la cellule, entraînant une toxicité cellulaire et une variété de symptômes neurologiques.
Les NLLD comprennent un certain nombre de maladies distinctes, notamment :
1. Maladie de Gaucher : causée par une déficience en glucocérébrosidase, entraînant une accumulation de glucosylcéramide dans les macrophages.
2. Maladie de Tay-Sachs : causée par une déficience en hexosaminidase A, entraînant une accumulation de gangliosides GM2 dans le cerveau.
3. Maladie de Niemann-Pick : causée par des déficiences en sphingomyélinases acide ou neutre, entraînant une accumulation de sphingomyéline et de cholestérol dans les cellules.
4. Maladie de Fabry : causée par une déficience en alpha-galactosidase A, entraînant une accumulation de globotriaosylcéramide (Gb3 ou GL-3) dans divers tissus.
5. Maladie de Sandhoff : causée par une déficience en hexosaminidase A et B, entraînant une accumulation de gangliosides GM2 dans le cerveau.
6. Maladie de Krabbe : causée par une déficience en galactocérébrosidase, entraînant une accumulation de psychosine dans le cerveau.
7. Maladie de Gaucher : causée par une déficience en glucocérébrosidase, entraînant une accumulation de glucosylcéramide dans les macrophages.
Les symptômes et la gravité des maladies varient considérablement, allant d'une forme légère à une forme grave et progressive qui peut entraîner une invalidité ou un décès prématuré. Les traitements actuels comprennent des thérapies de remplacement enzymatique, des médicaments modificateurs de la maladie et des soins de soutien pour gérer les symptômes.
L'acide lactique est un composé organique qui est produit dans les muscles pendant l'exercice intense ou lorsque les cellules ne reçoivent pas suffisamment d'oxygène pour décomposer le glucose en énergie. Ce processus, connu sous le nom de fermentation lactique, entraîne une accumulation d'acide lactique dans les muscles, ce qui peut provoquer des douleurs et une fatigue musculaires.
En outre, l'acide lactique est également produit en petites quantités par le corps en tout temps, même au repos, comme un sous-produit du métabolisme normal. Il est traité par le foie et excrété dans les urines.
Des niveaux élevés d'acide lactique dans le sang peuvent être le résultat d'une activité physique intense ou prolongée, de maladies cardiovasculaires, de diabète ou d'insuffisance hépatique. Des taux élevés d'acide lactique peuvent également indiquer une condition médicale grave appelée acidose lactique, qui est une accumulation excessive d'acide lactique dans le sang et peut être fatale si elle n'est pas traitée rapidement.
La maladie de surcharge en acide sialique est un trouble métabolique héréditaire rare caractérisé par l'accumulation excessive d'acide sialique dans l'organisme. L'acide sialique est une substance naturellement présente dans le corps et joue un rôle important dans la structure et la fonction des protéines et des lipides à la surface des cellules.
Cette maladie est causée par une mutation du gène SLC17A5, qui code pour un transporteur spécifique de l'acide sialique dans les lysosomes, où il est normalement dégradé. En conséquence, l'acide sialique s'accumule dans divers tissus et organes du corps, entraînant une variété de symptômes.
Les symptômes de la maladie de surcharge en acide sialique peuvent inclure des vomissements récurrents, une diarrhée sévère, une hypertrophie du foie et de la rate, une déficience intellectuelle, des convulsions, une ataxie (perte d'équilibre et de coordination), une neuropathie périphérique (dommages aux nerfs situés en dehors du cerveau et de la moelle épinière) et une vision floue.
Le diagnostic de cette maladie est généralement posé sur la base d'un examen clinique, d'une analyse biochimique des urines et d'une confirmation génétique. Le traitement actuel consiste principalement en des soins de soutien pour gérer les symptômes et améliorer la qualité de vie des patients. Des recherches sont en cours pour développer des thérapies spécifiques visant à réduire l'accumulation d'acide sialique dans le corps.
Le glucose est un monosaccharide simple, ou sucre simple, qui est la forme la plus fondamentale de sucre dans le métabolisme des glucides. Il s'agit d'un type d'aldohexose, ce qui signifie qu'il contient six atomes de carbone, un groupe aldéhyde et un groupe hydroxyle sur chaque atome de carbone à l'exception du premier et du dernier.
Le glucose est la principale source d'énergie pour les cellules vivantes, y compris les cellules humaines. Il est absorbé dans le sang après la digestion des glucides complexes ou des sucres simples contenus dans les aliments et fournit de l'énergie aux muscles et au cerveau.
Le taux de glucose sanguin (glycémie) est étroitement régulé par plusieurs hormones, dont l'insuline et le glucagon, pour maintenir un équilibre énergétique optimal dans le corps. Des niveaux anormalement élevés ou faibles de glucose peuvent indiquer divers troubles métaboliques, tels que le diabète sucré ou l'hypoglycémie.
Un dépendovirus est un type de virus qui nécessite l'expression d'au moins un gène spécifique dans les cellules hôtes pour assurer sa réplication et sa survie. Les dépendovirus appartiennent à la famille des *Parvoviridae* et sont définis par leur dépendance aux protéines de la famille des *US22* du virus herpès simplex (VHS) pour compléter leur cycle de réplication. Ces protéines sont exprimées dans les cellules infectées par le VHS, ce qui permet au dépendovirus d'utiliser ces ressources cellulaires pour se répliquer et produire de nouvelles particules virales.
Les dépendovirus ont un génome à simple brin d'ADN linéaire et sont incapables de s'intégrer dans le génome des cellules hôtes. Ils peuvent infecter une variété de types cellulaires, y compris les cellules humaines et animales. Bien que certains dépendovirus puissent causer des maladies chez l'homme, la plupart d'entre eux sont considérés comme étant à faible pathogénicité.
Les dépendovirus sont souvent utilisés en recherche biomédicale comme vecteurs pour transférer des gènes dans les cellules hôtes, car ils peuvent être modifiés génétiquement pour exprimer des gènes d'intérêt. Cela en fait un outil utile pour l'étude de la fonction des gènes et pour le développement de thérapies géniques.
La thérapie génétique est une forme avancée de médecine qui consiste à remplacer, manipuler ou inactiver des gènes spécifiques dans les cellules d'un patient pour traiter ou prévenir des maladies héréditaires ou acquises. Elle vise à corriger les défauts génétiques sous-jacents en introduisant des matériaux génétiques sains, tels que des gènes fonctionnels ou des acides nucléiques thérapeutiques, dans les cellules du patient.
Ces matériaux génétiques peuvent être délivrés directement aux cellules affectées par l'intermédiaire de vecteurs, tels que des virus inactivés ou des nanoparticules, qui permettent d'introduire les gènes thérapeutiques dans le génome ciblé. Une fois intégrés, ces nouveaux gènes peuvent aider à produire des protéines manquantes ou défectueuses, réguler l'expression de certains gènes, inhiber la production de protéines nocives ou même déclencher le processus de mort cellulaire programmée (apoptose) pour éliminer les cellules anormales.
Bien que la thérapie génétique offre des perspectives prometteuses dans le traitement de diverses affections, telles que les maladies génétiques rares, le cancer et certaines maladies infectieuses, elle est encore considérée comme une approche expérimentale et fait l'objet de recherches intensives pour évaluer son efficacité et sa sécurité à long terme.
Les muscles sont des organes contractiles qui forment une grande partie du tissu corporel. Ils sont responsables de la mobilité volontaire et involontaire dans le corps humain. Les muscles se contractent pour permettre le mouvement des os, aider à maintenir la posture et contribuer à diverses fonctions corporelles telles que la respiration, la digestion et la circulation sanguine.
Il existe trois types principaux de muscles dans le corps humain :
1. Les muscles squelettiques ou striés : Ils sont attachés aux os par des tendons et leur contraction permet les mouvements volontaires du corps. Ces muscles ont une apparence striée sous un microscope, d'où leur nom.
2. Les muscles lisses : Ce sont des muscles trouvés dans les parois des vaisseaux sanguins, des bronches, de l'utérus et du tube digestif. Ils fonctionnent involontairement, contrôlés par le système nerveux autonome, et participent à des fonctions telles que la circulation, la respiration et la digestion.
3. Le muscle cardiaque : C'est un type spécial de muscle strié qui forme la majeure partie du cœur. Il fonctionne automatiquement sans aucun contrôle volontaire, pompant le sang dans tout le corps.
La capacité des muscles à se contracter et à se détendre provient de leurs propriétés physiques uniques et de la présence de protéines spécialisées telles que l'actine et la myosine, qui glissent les unes contre les autres lorsque le muscle se contracte.
La Maladie de Wolman est une maladie héréditaire rare et grave, due à un déficit en lipase acide lysosomale, qui entraîne un accumulation excessive de cholestérol et de triglycérides dans différents organes du corps. Cela conduit à des dommages progressifs au foie, aux reins, aux intestins, aux glandes surrénales et au système squelettique. Les symptômes apparaissent généralement dès les premiers mois de vie et comprennent une augmentation du volume abdominal, des vomissements récurrents, une diarrhée sévère, une perte de poids et un retard de croissance. D'autres signes peuvent inclure une faiblesse musculaire, une anémie, une augmentation du taux de cholestérol sanguin et des calcifications dans les artères coronaires. La Maladie de Wolman est héréditaire et autosomique récessive, ce qui signifie qu'elle affecte les deux sexes et que les deux parents doivent être porteurs de la mutation génétique pour que l'enfant développe la maladie. Actuellement, il n'existe pas de traitement curatif pour cette maladie et le pronostic est généralement mauvais, avec une espérance de vie moyenne d'environ six mois.
Les lactates sont des ions hydrogénocarboxyliques qui se forment lorsque le lactate déshydrogénase convertit le pyruvate en acide lactique dans le processus métabolique connu sous le nom de glycolyse. Le lactate est produit dans les cellules lorsqu'il y a un manque d'oxygène ou une insuffisance d'oxydation du pyruvate par le cycle de Krebs dans la matrice mitochondriale.
Les lactates peuvent s'accumuler dans le sang, une condition appelée lactacidémie, en cas d'anomalies congénitales du métabolisme des acides ou d'une production accrue de lactate due à un apport insuffisant en oxygène (hypoxie) dans les tissus, comme lors d'un effort physique intense, d'une crise cardiaque, d'une insuffisance circulatoire ou d'une septicémie.
Des niveaux élevés de lactates peuvent indiquer un état métabolique anaérobie et sont associés à des maladies graves telles que l'insuffisance hépatique, l'insuffisance rénale, les lésions tissulaires, le choc septique et l'hypoxie. Les lactates sont également utilisés comme marqueurs de pronostic dans certaines affections médicales.
Les muscles squelettiques, également connus sous le nom de muscles striés squelettiques, sont des types spécifiques de tissus musculaires qui se connectent aux os et à d'autres structures via des tendons. Ils sont responsables de la production de force et de mouvements volontaires du corps. Les muscles squelettiques sont constitués de nombreuses fibres musculaires individuelles, organisées en faisceaux et recouvertes d'une membrane protectrice appelée épimysium. Chaque fibre musculaire est elle-même composée de plusieurs myofibrilles, qui contiennent des protéines contractiles telles que l'actine et la myosine. Ces protéines glissent les unes sur les autres lorsque le muscle se contracte, entraînant ainsi le mouvement des os auxquels elles sont attachées. Les muscles squelettiques peuvent également jouer un rôle dans la stabilisation articulaire, la posture et la thermorégulation du corps.
Le polymorphisme conformationnel simple brin (SSCP) est une technique de biologie moléculaire utilisée pour la détection de variations dans les séquences d'ADN. Il s'agit d'une méthode sensible pour la détection des mutations ponctuelles, y compris les substitutions et les petites insertions ou délétions (indels).
Dans cette technique, l'ADN cible est amplifié par PCR, puis le produit d'amplification est dénaturé en une seule chaîne. Cette chaîne simple est ensuite soumise à des conditions qui favorisent la formation de boucles et d'autres structures conformationnelles uniques pour chaque séquence d'ADN légèrement différente.
Lorsque les échantillons sont analysés par électrophorèse sur gel, les différentes conformations migreront à des vitesses différentes, ce qui permet de distinguer les variantes de la séquence d'intérêt. Les variations dans les modèles de migration peuvent indiquer la présence de mutations ponctuelles.
Le polymorphisme conformationnel simple brin est une méthode utile pour le dépistage des mutations dans les gènes associés à des maladies héréditaires, ainsi que pour la surveillance de l'évolution des tumeurs cancéreuses. Cependant, il peut être difficile d'interpréter les résultats de cette méthode en raison de la complexité des modèles de migration et de la possibilité de faux positifs ou négatifs. Par conséquent, il est souvent utilisé en combinaison avec d'autres techniques de séquençage pour confirmer les résultats.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
L'alpha-mannosidosis est une maladie rare du métabolisme des glycoprotéines, héréditaire et liée à l'X. Elle est causée par un déficit en alpha-mannosidase, une enzyme nécessaire à la dégradation des glycoprotéines dans les lysosomes. Lorsque cette enzyme fait défaut, les glycoprotéines ne peuvent pas être correctement décomposées et s'accumulent dans les cellules, entraînant une variété de symptômes.
Les symptômes de l'alpha-mannosidosis peuvent inclure des retards de développement, des problèmes d'élocution et de déglutition, des faciès caractéristiques, une perte auditive progressive, des problèmes cardiaques, des infections récurrentes, une augmentation de la taille du foie et de la rate, et dans les cas graves, une atteinte neurologique avec raideur musculaire, spasticité, convulsions et démence.
Le diagnostic de l'alpha-mannosidosis est généralement posé par des tests enzymatiques sur les leucocytes ou la fibroblaste cutanée. Le traitement peut inclure des soins de soutien pour gérer les symptômes, ainsi qu'une thérapie enzymatique substitutive expérimentale. Actuellement, il n'existe pas de cure pour cette maladie.
L'hypoxanthine est un produit chimique qui se forme dans le corps lors des processus de décomposition des purines, qui sont des substances présentes dans l'ADN et l'ARN. Elle joue également un rôle important dans la production d'énergie au niveau cellulaire.
Dans un contexte médical, les taux anormalement élevés d'hypoxanthine peuvent indiquer une affection sous-jacente, telle qu'une décomposition accélérée de purines due à des maladies génétiques rares telles que la goutte ou certaines formes d'anémie. Des niveaux élevés d'hypoxanthine peuvent également être observés dans le cerveau après un accident vasculaire cérébral, car les cellules cérébrales endommagées décomposent rapidement les purines pour produire de l'énergie.
Il est important de noter que des tests spécifiques sont nécessaires pour mesurer les niveaux d'hypoxanthine dans le corps, et ces tests ne sont généralement effectués que dans le cadre d'une enquête sur une affection sous-jacente suspectée.
L'acide urique est une substance chimique qui se forme lorsque votre corps décompose des purines, des substances présentes dans de nombreux aliments, comme la viande et les poissons. L'acide urique peut s'accumuler dans le sang et provoquer des cristaux douloureux dans les articulations et les tissus mous, entraînant une maladie appelée goutte.
L'acide urique est également présent dans l'urine et peut former des calculs rénaux s'il se cristallise dans les reins. Les facteurs de risque d'une accumulation excessive d'acide urique comprennent une consommation élevée d'alcool, certains médicaments, des maladies rénales et un régime alimentaire riche en viandes rouges, en fruits de mer et en alcool.
Il est important de maintenir des niveaux normaux d'acide urique dans le sang pour prévenir les complications telles que la goutte et les calculs rénaux. Cela peut être réalisé grâce à un mode de vie sain, y compris une alimentation équilibrée, une activité physique régulière et une consommation d'alcool modérée. Dans certains cas, des médicaments peuvent être prescrits pour réduire les niveaux d'acide urique dans le sang.
La glycémie est la concentration de glucose (un type de sucre) dans le sang. Elle est un indicateur crucial de la santé métabolique d'un individu. Le taux normal de glycémie à jeun devrait être entre 70 et 100 milligrammes par décilitre (mg/dL). Chez les personnes qui ne souffrent pas de diabète, après avoir mangé, la glycémie peut atteindre environ 140 mg/dL, mais elle redescend généralement à moins de 100 mg/dL deux heures plus tard. Des taux de glycémie constamment élevés peuvent indiquer un diabète ou une autre anomalie métabolique.
Un vecteur génétique est un outil utilisé en génétique moléculaire pour introduire des gènes ou des fragments d'ADN spécifiques dans des cellules cibles. Il s'agit généralement d'un agent viral ou bactérien modifié qui a été désarmé, de sorte qu'il ne peut plus causer de maladie, mais conserve sa capacité à infecter et à introduire son propre matériel génétique dans les cellules hôtes.
Les vecteurs génétiques sont couramment utilisés dans la recherche biomédicale pour étudier l'expression des gènes, la fonction des protéines et les mécanismes de régulation de l'expression génétique. Ils peuvent également être utilisés en thérapie génique pour introduire des gènes thérapeutiques dans des cellules humaines afin de traiter ou de prévenir des maladies causées par des mutations génétiques.
Les vecteurs viraux les plus couramment utilisés sont les virus adéno-associés (AAV), les virus lentiviraux et les rétrovirus. Les vecteurs bactériens comprennent les plasmides, qui sont des petites molécules d'ADN circulaires que les bactéries utilisent pour transférer du matériel génétique entre elles.
Il est important de noter que l'utilisation de vecteurs génétiques comporte certains risques, tels que l'insertion aléatoire de gènes dans le génome de l'hôte, ce qui peut entraîner des mutations indésirables ou la activation de gènes oncogéniques. Par conséquent, il est essentiel de mettre en place des protocoles de sécurité rigoureux pour minimiser ces risques et garantir l'innocuité des applications thérapeutiques des vecteurs génétiques.
Les glycogène synthase kinases (GSK) sont des enzymes qui jouent un rôle crucial dans la régulation du métabolisme des glucides. Plus spécifiquement, elles phosphorylent et inactivent la glycogène synthase, une enzyme clé dans le processus de glycogénogenèse, qui est la biosynthèse du glycogène à partir de glucose.
Il existe plusieurs isoformes de GSK, dont les plus étudiées sont GSK-3α et GSK-3β. Ces enzymes sont largement exprimées dans divers tissus, y compris le cerveau, le foie et le muscle squelettique. En plus de réguler la glycogénogenèse, les GSK sont également impliquées dans d'autres processus cellulaires tels que la signalisation cellulaire, la prolifération cellulaire, l'apoptose et la différentiation cellulaire.
L'activité des GSK est régulée par une variété de facteurs, y compris les hormones telles que l'insuline et le glucagon, ainsi que les voies de signalisation intracellulaire telles que la voie Wnt/β-caténine. Des anomalies dans l'activité des GSK ont été associées à un certain nombre de maladies, y compris le diabète sucré, la maladie d'Alzheimer et certains types de cancer.
Les sphingolipidoses sont un groupe de troubles héréditaires liés au métabolisme qui résultent d'un déficit en enzymes nécessaires pour décomposer les sphingolipides, une classe particulière de graisses complexes. Ces maladies comprennent la gangliosidose à GM1, la gangliosidose à GD1, la maladie de Gaucher, la maladie de Niemann-Pick, la maladie de Fabry, la maladie de Farber et la maladie de Krabbe.
Les sphingolipides s'accumulent dans les cellules du corps, en particulier dans le cerveau, entraînant une variété de symptômes qui peuvent inclure des anomalies neurologiques, des problèmes de développement, une hypertrophie du foie et de la rate, des anémies, des problèmes cutanés et des os fragiles. Le traitement peut inclure des thérapies enzymatiques de remplacement, des greffes de moelle osseuse et des stratégies de gestion des symptômes.
Je suis désolé, mais la phrase "Maladies Expérimentales" ne correspond pas à une définition médicale spécifique. Le terme "maladie expérimentale" peut faire référence à des études ou recherches scientifiques dans lesquelles des maladies sont simulées ou créées dans un laboratoire, généralement sur des animaux de laboratoire, pour mieux comprendre la pathogenèse, le développement et les traitements possibles d'une maladie. Cependant, il ne s'agit pas d'un terme médical standardisé. Si vous cherchez des informations sur une procédure ou un concept médical spécifique, veuillez me fournir plus de détails afin que je puisse vous aider au mieux.
Les données de séquence moléculaire se réfèrent aux informations génétiques ou protéomiques qui décrivent l'ordre des unités constitutives d'une molécule biologique spécifique. Dans le contexte de la génétique, cela peut inclure les séquences d'ADN ou d'ARN, qui sont composées d'une série de nucléotides (adénine, thymine, guanine et cytosine pour l'ADN; adénine, uracile, guanine et cytosine pour l'ARN). Dans le contexte de la protéomique, cela peut inclure la séquence d'acides aminés qui composent une protéine.
Ces données sont cruciales dans divers domaines de la recherche biologique et médicale, y compris la génétique, la biologie moléculaire, la médecine personnalisée, la pharmacologie et la pathologie. Elles peuvent aider à identifier des mutations ou des variations spécifiques qui peuvent être associées à des maladies particulières, à prédire la structure et la fonction des protéines, à développer de nouveaux médicaments ciblés, et à comprendre l'évolution et la diversité biologique.
Les technologies modernes telles que le séquençage de nouvelle génération (NGS) ont rendu possible l'acquisition rapide et économique de vastes quantités de données de séquence moléculaire, ce qui a révolutionné ces domaines de recherche. Cependant, l'interprétation et l'analyse de ces données restent un défi important, nécessitant des méthodes bioinformatiques sophistiquées et une expertise spécialisée.
Inosine est une substance chimique organique qui joue un rôle important dans le métabolisme des purines, un type de molécule présent dans l'ADN et l'ARN. Elle est produite dans l'organisme lorsque les cellules décomposent les purines en excès ou endommagées.
Inosine peut également être trouvée dans certains aliments, tels que la viande, le poisson et les noix. Dans le corps, inosine est convertie en hypoxanthine, une base nucléique qui peut être utilisée pour synthétiser de l'ADN et de l'ARN.
Inosine a également démontré des propriétés anti-inflammatoires et immunomodulatrices, ce qui en fait un sujet d'intérêt dans la recherche médicale pour le traitement de diverses maladies, y compris les maladies neurodégénératives et les lésions tissulaires. Cependant, des études supplémentaires sont nécessaires pour comprendre pleinement ses effets et son potentiel thérapeutique.
En génétique, un exon est une séquence d'ADN qui code pour une partie spécifique d'une protéine. Après la transcription de l'ADN en ARN messager (ARNm), les exons sont conservés et assemblés dans le processus de maturation de l'ARNm, tandis que les introns, qui sont les séquences non codantes, sont éliminés. Les exons forment ainsi la section codante finale de l'ARNm mature, qui est ensuite traduite en une chaîne polypeptidique lors de la synthèse des protéines.
En bref, un exon est une région d'un gène qui contribue à la séquence d'acides aminés d'une protéine après le traitement et l'assemblage de l'ARNm mature. Les mutations dans les exons peuvent entraîner des modifications dans la structure des protéines, ce qui peut conduire à des maladies génétiques ou à des changements phénotypiques.
Les lysosomes sont des organites membranaires trouvés dans la plupart des cellules eucaryotes. Ils jouent un rôle crucial dans le processus de dégradation et d'élimination des matières et des déchets cellulaires. Les lysosomes contiennent une variété d'enzymes hydrolytiques qui peuvent décomposer divers biomolécules telles que les lipides, les protéines, les glucides et les acides nucléiques en leurs composants constitutifs.
Les lysosomes sont souvent appelés «l'usine à ordures» de la cellule car ils aident à maintenir un environnement interne propre et sain en éliminant les déchets et les matières endommagées ou inutiles. Ils sont également impliqués dans le processus d'autophagie, dans lequel les composants cellulaires endommagés ou vieillissants sont encapsulés dans des membranes, formant une structure appelée autophagosome, qui fusionne ensuite avec un lysosome pour décomposer son contenu en nutriments réutilisables.
Les défauts de fonctionnement des lysosomes ont été associés à diverses maladies génétiques, telles que les maladies lysosomales, qui sont causées par des mutations dans les gènes codant pour les enzymes lysosomales ou d'autres protéines impliquées dans le fonctionnement des lysosomes. Ces maladies peuvent entraîner une accumulation de matériaux non dégradés dans la cellule, ce qui peut endommager les tissus et provoquer une variété de symptômes cliniques.
L'hypoxanthine est un composé organique qui appartient à la classe des purines. Dans le métabolisme des nucléotides, l'hypoxanthine est un produit intermédiaire de la dégradation de l'adénosine et de la guanosine. Elle peut être convertie en xanthine par l'action de l'enzyme hypoxanthine-guanine phosphoribosyltransférase, puis en urate par l'action de la xanthine oxydase. Des niveaux élevés d'hypoxanthine dans le sang peuvent indiquer une dégradation accrue des nucléotides, ce qui peut être observé dans certaines conditions pathologiques telles que les maladies hématologiques, les infections et l'insuffisance rénale. Cependant, il est important de noter que l'hypoxanthine n'est pas couramment utilisée comme marqueur clinique pour ces conditions.
Une souris knockout, également connue sous le nom de souris génétiquement modifiée à knockout, est un type de souris de laboratoire qui a eu un ou plusieurs gènes spécifiques désactivés ou "knockout". Cela est accompli en utilisant des techniques d'ingénierie génétique pour insérer une mutation dans le gène cible, ce qui entraîne l'interruption de sa fonction.
Les souris knockout sont largement utilisées dans la recherche biomédicale pour étudier les fonctions des gènes et leur rôle dans les processus physiologiques et pathologiques. En éliminant ou en désactivant un gène spécifique, les chercheurs peuvent observer les effets de cette perte sur le phénotype de la souris, ce qui peut fournir des informations précieuses sur la fonction du gène et ses interactions avec d'autres gènes et processus cellulaires.
Les souris knockout sont souvent utilisées dans l'étude des maladies humaines, car les souris partagent une grande similitude génétique avec les humains. En créant des souris knockout pour des gènes associés à certaines maladies humaines, les chercheurs peuvent étudier le rôle de ces gènes dans la maladie et tester de nouvelles thérapies potentielles.
Cependant, il est important de noter que les souris knockout ne sont pas simplement des modèles parfaits de maladies humaines, car elles peuvent présenter des différences dans la fonction et l'expression des gènes ainsi que dans les réponses aux traitements. Par conséquent, les résultats obtenus à partir des souris knockout doivent être interprétés avec prudence et validés dans d'autres systèmes de modèle ou dans des études cliniques humaines avant d'être appliqués à la pratique médicale.
Les maladies des chiens se réfèrent à un large éventail de conditions médicales qui peuvent affecter les chiens. Ces maladies peuvent être congénitales (présentes à la naissance), acquises (développées au cours de la vie du chien) ou infectieuses (causées par des agents pathogènes tels que des bactéries, des virus, des champignons ou des parasites).
Les maladies courantes chez les chiens comprennent les maladies de l'appareil digestif (comme la maladie inflammatoire de l'intestin et la pancréatite), les maladies cardiovasculaires (comme l'insuffisance cardiaque congestive et l'endocardite), les maladies respiratoires (comme la bronchite et la pneumonie), les maladies de la peau (comme la dermatite allergique et l'otite externe), les maladies du système nerveux (comme l'épilepsie et la maladie dégénérative du disque), les maladies des reins et des voies urinaires (comme l'insuffisance rénale et la cystite), le cancer et le diabète sucré.
Les chiens peuvent également être affectés par des maladies infectieuses telles que la rage, la parvovirose, la distemper, la leptospirose, la borréliose (maladie de Lyme) et l'hépatite contagieuse canine.
La prévention, le diagnostic et le traitement des maladies chez les chiens nécessitent une attention vétérinaire professionnelle. Les propriétaires de chiens doivent être attentifs aux signes de maladie, tels que la léthargie, la perte d'appétit, la vomissements, la diarrhée, la toux, les éternuements, les démangeaisons, les douleurs, les boiteries et les changements de comportement. Un diagnostic précoce et un traitement approprié peuvent aider à améliorer le pronostic et la qualité de vie des chiens atteints de maladies.
En génétique, un hétérozygote est un individu qui possède deux allèles différents d'un même gène sur les deux chromosomes homologues. Cela signifie que l'individu a hérité d'un allèle particulier du gène en question de chacun de ses parents, et ces deux allèles peuvent être différents l'un de l'autre.
Dans le contexte de la génétique mendélienne classique, un hétérozygote est représenté par une notation avec une lettre majuscule suivie d'un signe plus (+) pour indiquer que cet individu est hétérozygote pour ce gène spécifique. Par exemple, dans le cas d'un gène avec deux allèles A et a, un hétérozygote serait noté Aa.
La présence d'hétérozygotie peut entraîner des phénotypes variés, en fonction du type de gène concerné et de la nature des allèles en présence. Dans certains cas, l'allèle dominant (généralement représenté par une lettre majuscule) détermine le phénotype, tandis que dans d'autres cas, les deux allèles peuvent contribuer au phénotype de manière égale ou interactive.
Il est important de noter qu'être hétérozygote pour certains gènes peut conférer des avantages ou des inconvénients en termes de santé, de résistance aux maladies et d'autres caractéristiques. Par exemple, l'hétérozygotie pour certaines mutations associées à la mucoviscidose (fibrose kystique) peut offrir une protection contre certaines bactéries nocives de l'appareil respiratoire.
La maladie de Gaucher est un trouble héréditaire des lysosomes, qui sont des organites cellulaires responsables de la dégradation des molécules. Cette maladie est causée par une mutation du gène GBA qui code pour l'enzyme glucocérébrosidase. Lorsque cette enzyme fait défaut ou est insuffisamment active, une substance appelée glucosylcéramide s'accumule dans certaines cellules, principalement les macrophages.
Cette accumulation entraîne le développement de cellules anormales appelées «cellules de Gaucher», qui se caractérisent par leur grande taille et leurs inclusions riches en lipides. Ces cellules s'accumulent dans certains organes, notamment la rate, le foie et les os, entraînant une augmentation du volume de ces organes (splénomégalie, hépatomégalie), des anomalies osseuses et une déficience en hormones sexuelles.
La maladie de Gaucher se manifeste sous trois formes cliniques principales : la forme type 1, qui est la plus fréquente et se caractérise par une atteinte principalement viscérale sans atteinte neurologique ; la forme type 2, qui est une forme néonatale sévère avec atteinte neurologique rapide et fatale ; et la forme type 3, qui se caractérise par une atteinte neurologique progressive.
Le diagnostic de la maladie de Gaucher repose sur la mesure de l'activité de l'enzyme glucocérébrosidase dans les leucocytes ou dans les fibroblastes cutanés, ainsi que sur la détection de mutations du gène GBA. Le traitement de cette maladie repose sur la thérapie enzymatique substitutive, qui consiste à administrer une forme recombinante de l'enzyme glucocérébrosidase pour remplacer l'activité enzymatique déficiente.
Les tumeurs du foie sont des growths anormales qui se produisent dans cet organe. Ils peuvent être bénins (non cancéreux) ou malins (cancéreux).
Les tumeurs bénignes du foie comprennent les hémangiomes, les adénomes et les hyperplasies nodulaires focales. Ces types de tumeurs ne se propagent pas à d'autres parties du corps et peuvent souvent être surveillés sans traitement. Cependant, dans certains cas, ils peuvent causer des problèmes s'ils deviennent grands ou si leur croissance comprime les structures voisines.
Les tumeurs malignes du foie comprennent le carcinome hépatocellulaire (CHC) et les métastases hépatiques. Le CHC est une forme de cancer qui commence dans les cellules du foie, tandis que les métastases hépatiques sont des cancers qui se sont propagés au foie à partir d'autres parties du corps. Les deux types peuvent causer des dommages importants aux fonctions hépatiques et nécessitent un traitement agressif.
Les facteurs de risque pour le développement des tumeurs malignes du foie comprennent l'infection par le virus de l'hépatite B ou C, la consommation excessive d'alcool, l'obésité, le diabète et l'exposition à certains produits chimiques. Les symptômes des tumeurs du foie peuvent inclure une douleur ou une sensation de plénitude dans le haut de l'abdomen, une perte de poids inexpliquée, une jaunisse (jaunissement de la peau et du blanc des yeux), des nausées et des vomissements. Le diagnostic est généralement posé par imagerie médicale telle qu'une échographie ou une tomodensitométrie, suivie d'une biopsie pour confirmer le type de tumeur.
La mucopolysaccharidose de type VII, également connue sous le nom de syndrome de Sly, est un trouble héréditaire rare de stockage lysosomal. Il est causé par une mutation du gène GUSB qui code pour l'enzyme β-glucuronidase. Cette enzyme est responsable du métabolisme des glycosaminoglycanes (GAG), également appelés mucopolysaccharides. Lorsque cette enzyme fait défaut, les GAG s'accumulent dans les lysosomes des cellules, entraînant une variété de symptômes.
Les symptômes de la mucopolysaccharidose de type VII peuvent inclure une croissance et un développement anormaux, une taille et une tête anormalement grandes, une dysostose multiple (anomalies osseuses multiples), des problèmes cardiaques, des problèmes respiratoires, une hypertension artérielle intracrânienne, une hépatosplénomégalie (augmentation du volume du foie et de la rate), une hernie ombilicale ou inguinale, une déficience auditive, une vision floue due à une opacification de la cornée, et des faciès caractéristiques avec un front large, une dépression nasale, des pommettes proéminentes et une mâchoire prognathe.
Le diagnostic de la mucopolysaccharidose de type VII est généralement posé sur la base d'un examen clinique, d'une analyse d'urine pour détecter l'excès de GAG, et d'un test enzymatique pour mesurer l'activité de la β-glucuronidase dans les leucocytes ou les fibroblastes. Le traitement peut inclure une thérapie de remplacement enzymatique, des soins de support et une gestion des complications.
Une séquence nucléotidique est l'ordre spécifique et linéaire d'une série de nucléotides dans une molécule d'acide nucléique, comme l'ADN ou l'ARN. Chaque nucléotide se compose d'un sucre (désoxyribose dans le cas de l'ADN et ribose dans le cas de l'ARN), d'un groupe phosphate et d'une base azotée. Les bases azotées peuvent être adénine (A), guanine (G), cytosine (C) et thymine (T) dans l'ADN, tandis que dans l'ARN, la thymine est remplacée par l'uracile (U).
La séquence nucléotidique d'une molécule d'ADN ou d'ARN contient des informations génétiques cruciales qui déterminent les caractéristiques et les fonctions de tous les organismes vivants. La décodage de ces séquences, appelée génomique, est essentiel pour comprendre la biologie moléculaire, la médecine et la recherche biologique en général.
En termes médicaux, la généalogie est l'étude systématique des antécédents familiaux et médicaux d'une personne ou d'une famille sur plusieurs générations. Elle vise à identifier les modèles de maladies héréditaires ou génétiques dans une famille, ce qui peut aider à évaluer le risque de développer certaines affections et à mettre en œuvre des stratégies de prévention et de dépistage appropriées.
Les professionnels de la santé utilisent souvent des arbres généalogiques pour représenter visuellement les relations familiales et les antécédents médicaux. Ces outils peuvent être particulièrement utiles dans la pratique clinique, en particulier lorsqu'il s'agit de maladies rares ou complexes qui ont tendance à se produire dans certaines familles en raison de facteurs génétiques sous-jacents.
En plus d'être un outil important pour la médecine préventive, la généalogie peut également fournir des informations précieuses sur l'histoire naturelle de diverses maladies et conditions, ce qui contribue à faire progresser notre compréhension globale de la pathogenèse et de la physiopathologie. Par conséquent, elle joue un rôle crucial dans la recherche médicale et les soins cliniques.
Les erreurs innées du métabolisme lipidique sont un groupe de troubles génétiques qui affectent la capacité du corps à traiter et à décomposer les graisses correctement. Ces maladies sont causées par des mutations dans les gènes qui codent pour les enzymes ou les protéines nécessaires au métabolisme des lipides.
Les lipides, également appelés graisses, sont des molécules importantes qui fournissent de l'énergie, construisent les membranes cellulaires et participent à la signalisation cellulaire. Dans les erreurs innées du métabolisme lipidique, le processus de décomposition et d'utilisation des graisses est altéré, entraînant une accumulation toxique de graisses ou de leurs produits intermédiaires dans divers tissus corporels.
Les symptômes et la gravité de ces maladies varient considérablement en fonction du type d'erreur innée du métabolisme lipidique et de l'ampleur de l'activité résiduelle de l'enzyme affectée. Les symptômes courants peuvent inclure une faiblesse musculaire, des crises, des problèmes neurologiques, une croissance ralentie, une hépatomégalie (augmentation du volume du foie), une splénomégalie (augmentation du volume de la rate) et une déficience intellectuelle.
Les exemples courants d'erreurs innées du métabolisme lipidique comprennent les acides gras à chaîne très longue (AGVL) bêta-oxydation, les maladies périoxysomales, la maladie de Gaucher, la maladie de Niemann-Pick et la maladie de Fabry. Le diagnostic de ces maladies est généralement établi par des tests génétiques et des analyses d'activité enzymatique spécifiques.
Le traitement des erreurs innées du métabolisme lipidique peut inclure un régime alimentaire spécial, une supplémentation en nutriments, des médicaments pour contrôler les symptômes et, dans certains cas, des thérapies enzymatiques substitutives. La prise en charge précoce et agressive de ces maladies peut aider à améliorer les résultats et la qualité de vie des patients.
Une mutation ponctuelle est un type spécifique de mutation génétique qui implique l'alteration d'une seule paire de bases dans une séquence d'ADN. Cela peut entraîner la substitution, l'insertion ou la délétion d'un nucléotide, ce qui peut à son tour modifier l'acide aminé codé par cette région particulière de l'ADN. Si cette modification se produit dans une région codante d'un gène (exon), cela peut entraîner la production d'une protéine anormale ou non fonctionnelle. Les mutations ponctuelles peuvent être héréditaires, transmises de parents à enfants, ou spontanées, survenant de novo dans un individu. Elles sont souvent associées à des maladies génétiques, certaines formes de cancer et au vieillissement.
Une mutation « faux sens » (ou missense mutation) est un type de mutation génétique où une seule paire de bases dans l'ADN est modifiée, ce qui entraîne le remplacement d'un acide aminé par un autre dans la protéine codée par ce gène. Cela peut altérer la fonction, la structure ou la stabilité de la protéine, dépendant de la position et de l'importance de l'acide aminé remplacé. Dans certains cas, ces mutations peuvent entraîner des maladies génétiques ou prédisposer à certaines conditions médicales. Toutefois, il est important de noter que toutes les mutations faux sens ne sont pas nécessairement pathogènes et que leur impact sur la santé dépend du contexte dans lequel elles se produisent.
La mucopolysaccharidose de type I (MPS I) est une maladie héréditaire rare associée à un déficit en alpha-L-iduronidase, une enzyme nécessaire pour dégrader des grandes molécules appelées glycosaminoglycanes (GAG). Lorsque cette enzyme fait défaut, les GAG s'accumulent dans les cellules, entraînant une variété de symptômes.
Il existe trois formes cliniques distinctes de MPS I : la forme sévère ou Hurler, la forme intermédiaire ou Hurler-Scheie et la forme légère ou Scheie. Les manifestations cliniques varient en fonction du type, mais peuvent inclure des traits dysmorphiques faciaux, une augmentation de la taille de la rate et/ou du foie, des problèmes cardiovasculaires, des difficultés respiratoires, une déficience auditive, une atteinte oculaire, une raideur articulaire, une déformation osseuse et un retard mental ou psychomoteur.
Le diagnostic de MPS I repose généralement sur l'analyse d'un échantillon de sang pour mesurer l'activité de l'enzyme alpha-L-iduronidase et confirmer la présence de mutations génétiques spécifiques à cette maladie. Le traitement peut inclure une thérapie enzymatique substitutive, des soins de support et des interventions chirurgicales pour gérer les complications associées à la maladie.
En médecine, la conservation d'un médicament fait référence à l'action ou au processus de préserver et de maintenir la stabilité, la qualité, l'efficacité et la sécurité d'un médicament pendant une certaine période. Cela implique généralement le stockage adéquat du médicament dans des conditions spécifiques telles qu'une température et une humidité contrôlées, à l'abri de la lumière directe du soleil et de tout dommage mécanique.
La conservation adéquate d'un médicament est essentielle pour garantir sa qualité et son efficacité lorsqu'il est utilisé chez les patients. Les fabricants de médicaments doivent tester la stabilité de leurs produits dans diverses conditions de stockage et spécifier des instructions de conservation appropriées sur l'étiquette du produit.
Il est important de suivre ces instructions de conservation pour assurer la sécurité et l'efficacité continues du médicament. Si un médicament n'est pas conservé correctement, cela peut entraîner une dégradation ou une altération de sa composition chimique, ce qui peut affecter son efficacité thérapeutique ou même entraîner des effets indésirables imprévus.
En résumé, la conservation médicamenteuse est un aspect crucial de la pratique médicale et pharmaceutique, qui vise à préserver la qualité, l'efficacité et la sécurité des médicaments pour une utilisation optimale chez les patients.
Je suis désolé, "chiens" est un terme général qui fait référence à une espèce spécifique d'animaux domestiques et non à une condition ou à un état médical. Les chiens (Canis lupus familiaris) sont des mammifères carnivores de la famille des Canidae. Ils sont souvent considérés comme les plus anciens animaux domestiqués par l'homme, depuis probablement 20 000 à 40 000 ans. Les chiens existent en plusieurs races, tailles et formes, avec des variations significatives dans leur apparence, leur tempérament et leurs capacités. Ils sont souvent élevés comme animaux de compagnie en raison de leur loyauté, de leur intelligence et de leur capacité à être formés.
Si vous cherchez une définition médicale ou des informations sur un sujet spécifique, veuillez me fournir plus de détails afin que je puisse vous aider au mieux.
La neutropénie est un terme médical qui décrit une condition où le nombre de neutrophiles dans le sang est considérablement réduit. Les neutrophiles sont un type de globules blancs (leucocytes) qui jouent un rôle crucial dans la défense du corps contre les infections, en particulier celles causées par des bactéries et des champignons.
Une personne est considérée comme neutropénique lorsque le nombre de neutrophiles dans son sang tombe en dessous d'un certain seuil, généralement défini comme moins de 1500 cellules par microlitre de sang. Cependant, les risques d'infections augmentent considérablement lorsque ce nombre chute à moins de 500 cellules par microlitre.
La neutropénie peut être classée comme légère, modérée ou sévère en fonction du nombre de neutrophiles dans le sang :
- Légère : entre 1000 et 1499 cellules/µL
- Modérée : entre 500 et 999 cellules/µL
- Sévère : moins de 500 cellules/µL
Cette condition peut être temporaire ou chronique et peut être causée par divers facteurs, tels que des infections virales, certains médicaments (comme la chimiothérapie), des maladies auto-immunes, des troubles hématologiques ou une carence en vitamine B12 et acide folique. La neutropénie peut entraîner un risque accru d'infections graves, voire mortelles, si elle n'est pas détectée et traitée rapidement.
Une séquence d'acides aminés est une liste ordonnée d'acides aminés qui forment une chaîne polypeptidique dans une protéine. Chaque protéine a sa propre séquence unique d'acides aminés, qui est déterminée par la séquence de nucléotides dans l'ADN qui code pour cette protéine. La séquence des acides aminés est cruciale pour la structure et la fonction d'une protéine. Les différences dans les séquences d'acides aminés peuvent entraîner des différences importantes dans les propriétés de deux protéines, telles que leur activité enzymatique, leur stabilité thermique ou leur interaction avec d'autres molécules. La détermination de la séquence d'acides aminés d'une protéine est une étape clé dans l'étude de sa structure et de sa fonction.
Un adénome est un type de tumeur non cancéreuse (bénigne) qui se développe dans les glandes. Il peut se former dans divers endroits du corps où il y a des glandes, mais ils sont le plus souvent trouvés dans la prostate, les glandes surrénales et les glandes hypophysaires. Les adénomes sont généralement lents à se développer et ne se propagent pas à d'autres parties du corps. Cependant, selon leur taille et leur emplacement, ils peuvent causer des problèmes de santé en comprimant les tissus voisins ou en interférant avec leur fonction normale.
Les adénomes peuvent ne pas provoquer de symptômes, surtout s'ils sont petits. Cependant, selon l'emplacement et la taille de la tumeur, des symptômes peuvent apparaître. Par exemple, un adénome de la prostate peut causer des problèmes de miction, tandis qu'un adénome de la glande pituitaire peut entraîner une production excessive d'hormones ou une vision floue.
Le traitement dépend de la taille et de l'emplacement de la tumeur, ainsi que des symptômes qu'elle provoque. Dans certains cas, aucun traitement n'est nécessaire et la tumeur est simplement surveillée. Dans d'autres cas, une intervention chirurgicale ou une radiothérapie peut être recommandée pour enlever la tumeur ou réduire sa taille.
Les microsomes sont des structures membranaires fragmentées qui se trouvent dans le réticulum endoplasmique rugueux (RER) des cellules. Ils sont souvent décrits comme les fragments vésiculaires du RER après la rupture de ses membranes pendant le processus de préparation d'échantillons en biologie cellulaire et en biochimie. Les microsomes contiennent des ribosomes liés à leur surface, ce qui leur permet de jouer un rôle crucial dans la synthèse des protéines.
De plus, les microsomes hébergent également plusieurs enzymes importantes, y compris le système du cytochrome P450, qui est responsable du métabolisme d'un large éventail de substances endogènes et exogènes, telles que les médicaments, les toxines et les hormones stéroïdes. Par conséquent, les microsomes sont souvent utilisés dans les études pour comprendre le métabolisme des xénobiotiques et l'effet des médicaments sur différents tissus corporels.
Il est important de noter que les microsomes ne sont pas présents en tant qu'organites distincts dans les cellules vivantes, mais plutôt comme des fragments de membranes produits lors des procédures de laboratoire.
Les lipidoses sont un groupe de maladies métaboliques héréditaires caractérisées par l'accumulation anormale de certaines graisses, appelées lipides, dans divers tissus corporels. Ces affections résultent généralement d'un déficit en une enzyme spécifique nécessaire au métabolisme des lipides.
Il existe plusieurs types de lipidoses, dont la plus courante est la maladie de Gaucher. Dans cette maladie, l'enzyme glucocérébrosidase fait défaut, entraînant l'accumulation de glucosylcéramide dans les macrophages, appelés cellules de Gaucher. Cela peut provoquer une augmentation du volume des organes affectés, tels que la rate et le foie, ainsi qu'une anémie et une thrombocytopénie.
D'autres types de lipidoses comprennent la maladie de Niemann-Pick, la maladie de Fabry, la gangliosidose à GM1 et la neuraminide O. Chacune de ces affections est causée par un déficit en une enzyme spécifique, entraînant l'accumulation d'un certain lipide dans divers tissus corporels.
Les symptômes des lipidoses peuvent varier considérablement en fonction du type et de la gravité de la maladie. Ils peuvent inclure une hypertrophie du foie et de la rate, une anémie, une thrombocytopénie, des problèmes neurologiques, des difficultés respiratoires, des anomalies cutanées et des problèmes de vision.
Le traitement des lipidoses dépend du type et de la gravité de la maladie. Dans certains cas, un traitement enzymatique de remplacement peut être utilisé pour remplacer l'enzyme manquante et ralentir ou arrêter l'accumulation de lipides. D'autres traitements peuvent inclure des médicaments pour gérer les symptômes, une thérapie de support et une gestion des complications.
L'analyse des mutations de l'ADN est une méthode d'examen génétique qui consiste à rechercher des modifications (mutations) dans la séquence de l'acide désoxyribonucléique (ADN). L'ADN est le matériel génétique présent dans les cellules de tous les organismes vivants et contient les instructions pour le développement, la fonction et la reproduction des organismes.
Les mutations peuvent survenir spontanément ou être héritées des parents d'un individu. Elles peuvent entraîner des changements dans la structure et la fonction des protéines, ce qui peut à son tour entraîner une variété de conséquences, allant de mineures à graves.
L'analyse des mutations de l'ADN est utilisée dans un large éventail d'applications, y compris le diagnostic et le suivi des maladies génétiques, la détermination de la susceptibilité à certaines maladies, l'identification des auteurs de crimes, la recherche sur les maladies et le développement de médicaments.
Il existe différentes méthodes pour analyser les mutations de l'ADN, notamment la séquençage de nouvelle génération (NGS), la PCR en temps réel, la PCR quantitative et la Southern blotting. Le choix de la méthode dépend du type de mutation recherchée, de la complexité du test et des besoins du patient ou du chercheur.
Adenoviridae est une famille de virus qui comprend plus de 50 types différents qui peuvent causer des infections chez les humains et d'autres animaux. Ces virus sont nommés d'après le tissu lymphoïde où ils ont été initialement isolés, à savoir les glandes adénoïdes.
Les adénovirus humains peuvent causer une variété de maladies, notamment des infections respiratoires hautes et basses, des conjonctivites, des gastro-entérites, des cystites interstitielles, et des infections du système nerveux central. Les symptômes dépendent du type de virus et peuvent varier d'une infection légère à une maladie grave.
Les adénovirus sont transmis par contact direct avec les sécrétions respiratoires ou fécales d'une personne infectée, ainsi que par contact avec des surfaces contaminées. Ils peuvent également être transmis par voie aérienne lorsqu'une personne infectée tousse ou éternue.
Les adénovirus sont résistants à la chaleur et au dessèchement, ce qui les rend difficiles à éliminer de l'environnement. Ils peuvent survivre pendant de longues périodes sur des surfaces inanimées, telles que les poignées de porte, les téléphones et les jouets.
Actuellement, il n'existe pas de vaccin disponible pour prévenir toutes les infections à adénovirus. Cependant, un vaccin contre certains types d'adénovirus est utilisé pour protéger les militaires en bonne santé contre les infections respiratoires aiguës. Les mesures de prévention comprennent le lavage des mains régulier, l'évitement du contact étroit avec une personne malade et le nettoyage et la désinfection des surfaces contaminées.
Les purines sont des bases azotées qui forment, avec les pyrimidines, la structure moléculaire des acides nucléiques, tels que l'ADN et l'ARN. Les purines comprennent l'adénine (A) et la guanine (G). Elles jouent un rôle crucial dans la synthèse de l'ADN, l'ARN, l'ATP (adénosine triphosphate), les coenzymes, ainsi que dans divers processus métaboliques. Les purines sont dégradées en acide urique, qui est excrété dans l'urine. Des taux élevés d'acide urique peuvent entraîner des maladies telles que la goutte et la néphrolithiase (calculs rénaux).
L'insuline est une hormone essentielle produite par les cellules bêta du pancréas. Elle joue un rôle crucial dans le métabolisme des glucides, des lipides et des protéines en régulant le taux de sucre dans le sang (glucose sanguin). Après avoir mangé, lorsque la glycémie augmente, l'insuline est libérée pour permettre aux cellules du corps d'absorber le glucose et l'utiliser comme source d'énergie ou de le stocker sous forme de glycogène dans le foie et les muscles. L'insuline favorise également la synthèse des protéines et des lipides à partir du glucose.
Dans certaines conditions, telles que le diabète sucré, la production ou l'action de l'insuline peut être altérée, entraînant une hyperglycémie (taux élevé de sucre dans le sang). Les personnes atteintes de diabète de type 1 doivent recevoir des injections d'insuline pour remplacer l'hormone manquante, tandis que les personnes atteintes de diabète de type 2 peuvent être traitées par des modifications du mode de vie, des médicaments oraux ou une insulinothérapie dans certains cas.
La fucosidose est une maladie héréditaire rare et grave qui appartient au groupe des maladies lysosomales. Elle est causée par un déficit en activité de l'enzyme alpha-L-fucosidase, ce qui entraîne l'accumulation de substrats dans les lysosomes et endommage progressivement les cellules du corps.
Cette maladie affecte principalement le cerveau, la moelle osseuse, le foie, la rate et le système nerveux périphérique. Les symptômes peuvent inclure des retards de développement, des problèmes moteurs, une hypertrophie du foie et de la rate, des anomalies faciales, des convulsions, des problèmes cardiaques et une déficience auditive.
La fucosidose est héritée de manière autosomique récessive, ce qui signifie que les deux copies du gène doivent être anormales pour que la maladie se développe. Les personnes atteintes de cette maladie ont généralement une espérance de vie courte et il n'existe actuellement aucun traitement curatif pour cette maladie.
 Glycogénose type 3 - Wikipedia
Glycogénose type 3 - Wikipedia
 Glycogénose de type III - Institut de Myologie
Glycogénose de type III - Institut de Myologie
 Traitement - L'Association Francophone des Glycogénoses
Traitement - L'Association Francophone des Glycogénoses
Les actions
 Glycogénose de type IIIa chez le chien | C.H.V Fregis - Fregis
Glycogénose de type IIIa chez le chien | C.H.V Fregis - Fregis
 Haute Autorité de Santé - Résultat de recherche
Haute Autorité de Santé - Résultat de recherche
 Maladies de surcharge glycogénique (glycogénoses) - Pédiatrie - Édition professionnelle du Manuel MSD
Maladies de surcharge glycogénique (glycogénoses) - Pédiatrie - Édition professionnelle du Manuel MSD
métabolisme - CISMeF
 28 Février 2013 : l'actualité au féminin
- Terrafemina
28 Février 2013 : l'actualité au féminin
- Terrafemina
 theses.fr - Julien Calderaro , Caractérisation moléculaire des adénomes hépatocytaires et des lésions prénéoplasiques hépatiques
theses.fr - Julien Calderaro , Caractérisation moléculaire des adénomes hépatocytaires et des lésions prénéoplasiques hépatiques
 Sources scientifiques - Vaincre les Maladies Lysosomales
Sources scientifiques - Vaincre les Maladies Lysosomales
 I-Stem - Équipe de Recherche : Dystrophies Musculaires (LGMDs)
I-Stem - Équipe de Recherche : Dystrophies Musculaires (LGMDs)
 InfoCancer - ARCAGY - GINECO - Les localisations - Cancers de l'enfant - Oncopédiatrie - L'hépatoblastome - PRETEXT - maladie...
InfoCancer - ARCAGY - GINECO - Les localisations - Cancers de l'enfant - Oncopédiatrie - L'hépatoblastome - PRETEXT - maladie...
 Localisations
Localisations
 Des Coon's Holidays - Élevage de Maine Coon à Reims
Des Coon's Holidays - Élevage de Maine Coon à Reims
 Métabolisme. TP Glucose 6-phosphatase (néoglucogenèse), QCM
Métabolisme. TP Glucose 6-phosphatase (néoglucogenèse), QCM
 Stand up paddle passion, le web magazine du sup et du paddle.
Stand up paddle passion, le web magazine du sup et du paddle.
 CHU-ANGERS - Médecine interne - Immunologie clinique
CHU-ANGERS - Médecine interne - Immunologie clinique
CHU-ANGERS - Médecine interne - Immunologie clinique
 Alimentation thérapeutique - Mar Bordanova
Alimentation thérapeutique - Mar Bordanova
 La stéatose hépatique - Nutrition & pédiatrie
La stéatose hépatique - Nutrition & pédiatrie
 Téléthon 2023 : date, don, le nom du parrain révélé
Téléthon 2023 : date, don, le nom du parrain révélé
Listes de discussion - lists
 EXTRANEAL, solution pour dialyse péritonéale
EXTRANEAL, solution pour dialyse péritonéale
 Troubles du cycle de l'urée (UCD) | Vitaflo France
Troubles du cycle de l'urée (UCD) | Vitaflo France
Analyses - Liste filtrée des analyses
Gen&Zic
Maladie7
- La glycogénose type III est une maladie génétique du métabolisme des glucides de la famille des glycogénoses qui se manifeste par une carence en amylo-1,6-glucocidase, l'enzyme débranchante (en) du glycogène. (wikipedia.org)
- La glycogénose de type III est une maladie à transmission autosomique récessive. (wikipedia.org)
- Le rôle de L'Association Francophone des Glycogénoses est d'être un pôle d'entraide pour toutes les personnes concernées par cette maladie et de promouvoir la recherche et l'effort médical. (glycogenoses.org)
- Découvrez la glycogénose de type IIIa, une maladie métabolique rare qui peut affecter certains chiens. (fregis.com)
- La glycogénose de type IIIa (maladie de Cori) chez le chien est une maladie lysosomale du jeune chien due au mauvais fonctionnement d'une enzyme (glycogen debranching enzyme : GDE) provoquant alors une accumulation anormale de glycogène dans le foie et les muscles. (fregis.com)
- La glycogénose de type I (GSD I) est une maladie métabolique héréditaire rare qui prédispose au développement des AHC. (theses.fr)
- La maladie de Danon est une glycogénose lysosomale due au déficit en LAMP-2 (Lysosomal-Associated Membrane Protein 2). (vml-asso.org)
Maladies3
- Le pronostic et le traitement des maladies de surcharge glycogénique varient selon le type, mais le traitement comprend généralement une supplémentation diététique avec de la fécule de maïs afin d'apporter une source durable de glucose pour les formes hépatiques de glycogénose et l'abstention d'exercices physiques dans les formes musculaires. (msdmanuals.com)
- Le labo U1213 "Nutrition, diabète et cerveau" de l'Inserm à Lyon , c'est une petite quinzaine de personnes travaillant sur deux « maladies miroirs » : le diabète de type 2, caractérisé par une hyperglycémie, et la glycogénose de type 1, qui provoque de dangereuses hypoglycémies. (genetzic.com)
- D'autres maladies du foie peuvent également évoluer en cancer, notamment les glycogénoses de type I et III, les porphyries aiguës ou la tyrosinémie. (gastroenterologie-paris.com)
Type3
- Il n'existe pas de traitement de la glycogénose de type IIIa. (fregis.com)
- Un petit garçon de 6 ans a mené un projet fort ambitieux pour venir en aide à son meilleur ami atteint d'une Glycogénose de type 1B. (terrafemina.com)
- Le diagnostic différentiel inclut la myopathie vacuolaire avec autophagie excessive liée à l'X (XMEA) et la glycogénose de type II (voir ces termes). (vml-asso.org)
Spinale1
- Nous avons de nombreux reproducteurs, tous inscrits au Livre Officiel des Origines Félines (LOOF) et déclarés sains suite aux tests contre la déficience en pyruvate kinase (PKdef), l'atrophie musculaire spinale (SMA), l'hypertrophie myocardique (HCM), la glycogénose (GS), le virus de l'immunodéficience féline (FIV) ainsi que la leucose féline due au FIV (FeLV). (chatsdumonde.com)
Personnes1
- Au quotidien, l'AFG constituée exclusivement de bénévoles, échange avec des personnes atteintes de glycogénose, leurs familles, des donateurs, des partenaires et bien d'autres. (glycogenoses.org)
Traitement1
- Les termes « AFG », « nous », « notre » et « nos » désignent l'Association Francophone des Glycogénoses en tant que responsable du traitement de vos données à caractère personnel, sauf indication contraire dans la présente Politique de confidentialité. (glycogenoses.org)