Hexosaminidases
beta-N-Acetylhexosaminidases
Hexosaminidase B
Lipidoses
Acetylglucosaminidases
Les hexosaminidases sont des enzymes hydrolases qui catalysent la réaction de clivage des glycosides d'hexosamine, libérant un sucre et un résidu aglycone. Il existe plusieurs types d'hexosaminidases, mais les deux plus étudiées sont l'hexosaminidase A et l'hexosaminidase B.
L'hexosaminidase A est responsable du clivage des glycosides de N-acétylglucosamine et de N-acétylgalactosamine, tandis que l'hexosaminidase B clive principalement les glycosides de N-acétylglucosamine.
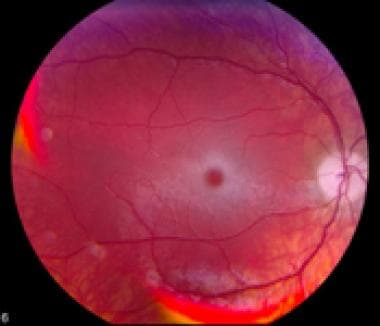
Les déficits en hexosaminidases A et B sont associés à des maladies héréditaires graves telles que la maladie de Tay-Sachs et la maladie de Sandhoff, respectivement. Ces maladies sont caractérisées par l'accumulation de gangliosides dans les neurones du cerveau, entraînant une dégénérescence neurologique progressive et souvent fatale.
Les β-N-acétylhexosaminidases sont des enzymes qui catalysent la dégradation des glycosphingolipides, en particulier les gangliosides, dans les lysosomes. Ces enzymes clivent le lien entre deux sucres, la N-acétylglucosamine et la galactose ou la N-acétylneuraminique, dans la molécule de glycosphingolipide.
Il existe trois isoformes de cette enzyme chez l'homme : α/β, β/β et α/α. Les mutations dans le gène codant pour les sous-unités α et β peuvent entraîner des maladies héréditaires graves telles que la maladie de Tay-Sachs et la maladie de Sandhoff, respectivement. Ces maladies sont caractérisées par l'accumulation de glycosphingolipides dans les neurones, entraînant une dégénérescence neurologique progressive.
En plus de leur rôle dans la dégradation des glycosphingolipides, les β-N-acétylhexosaminidases sont également importantes pour la régulation de la signalisation cellulaire et l'inflammation.
La hexosaminidase B est une enzyme essentielle dans le corps humain qui joue un rôle crucial dans le métabolisme des glycosphingolipides. Plus spécifiquement, elle est responsable de la dégradation du ganglioside GM2 en composants plus simples. Ce processus aide à prévenir l'accumulation toxique de ces lipides dans les cellules, en particulier dans les neurones.
Une déficience en hexosaminidase B entraîne une maladie génétique rare appelée maladie de Tay-Sachs, qui se caractérise par l'accumulation de gangliosides GM2 dans le cerveau et la moelle épinière. Cela peut conduire à une dégénérescence neurologique grave et progressive, entraînant souvent une invalidité et une mort prématurée.
Il est important de noter que l'activité de la hexosaminidase B est souvent utilisée comme marqueur diagnostique pour détecter la présence de cette maladie. Des niveaux anormalement bas d'hexosaminidase B peuvent indiquer une suspicion de maladie de Tay-Sachs, bien que des tests génétiques supplémentaires soient nécessaires pour confirmer le diagnostic.
Les lipidoses sont un groupe de maladies métaboliques héréditaires caractérisées par l'accumulation anormale de certaines graisses, appelées lipides, dans divers tissus corporels. Ces affections résultent généralement d'un déficit en une enzyme spécifique nécessaire au métabolisme des lipides.
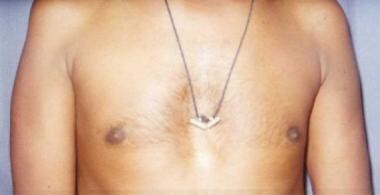
Il existe plusieurs types de lipidoses, dont la plus courante est la maladie de Gaucher. Dans cette maladie, l'enzyme glucocérébrosidase fait défaut, entraînant l'accumulation de glucosylcéramide dans les macrophages, appelés cellules de Gaucher. Cela peut provoquer une augmentation du volume des organes affectés, tels que la rate et le foie, ainsi qu'une anémie et une thrombocytopénie.
D'autres types de lipidoses comprennent la maladie de Niemann-Pick, la maladie de Fabry, la gangliosidose à GM1 et la neuraminide O. Chacune de ces affections est causée par un déficit en une enzyme spécifique, entraînant l'accumulation d'un certain lipide dans divers tissus corporels.
Les symptômes des lipidoses peuvent varier considérablement en fonction du type et de la gravité de la maladie. Ils peuvent inclure une hypertrophie du foie et de la rate, une anémie, une thrombocytopénie, des problèmes neurologiques, des difficultés respiratoires, des anomalies cutanées et des problèmes de vision.
Le traitement des lipidoses dépend du type et de la gravité de la maladie. Dans certains cas, un traitement enzymatique de remplacement peut être utilisé pour remplacer l'enzyme manquante et ralentir ou arrêter l'accumulation de lipides. D'autres traitements peuvent inclure des médicaments pour gérer les symptômes, une thérapie de support et une gestion des complications.
Les acetylglucosaminidases sont des enzymes qui dégradent et catalysent la réaction d'hydrolyse des glycosides spécifiques, tels que les N-acétylglucosamine liés aux protéines ou aux lipides. Ces enzymes jouent un rôle important dans le métabolisme des oligosaccharides et des glycoprotéines, ainsi que dans la biosynthèse et la dégradation des glycosaminoglycanes et des gangliosides. Les défauts de ces enzymes peuvent entraîner diverses maladies génétiques, telles que les mucolipidoses et les gangliosidoses.
Il existe plusieurs types d'acetylglucosaminidases, chacune avec des spécificités différentes pour leurs substrats. Par exemple, l'acetylglucosaminidase acide est une enzyme qui se trouve principalement dans les lysosomes et dégrade les N-acétylglucosamine liés aux protéines. D'autres types d'acetylglucosaminidases peuvent être trouvés dans le réticulum endoplasmique, où ils jouent un rôle dans la modification des glycoprotéines.
Les acetylglucosaminidases sont également utilisées à des fins de diagnostic et de thérapie. Par exemple, l'activité de ces enzymes peut être mesurée pour diagnostiquer certaines maladies génétiques, telles que la maladie de Tay-Sachs ou la gangliosidose à GM1. De plus, des substituts d'enzymes recombinantes peuvent être utilisés comme thérapie enzymatique substitutive pour traiter certaines de ces maladies.
 Hexosaminidase - Wikipedia
Hexosaminidase - Wikipedia Misoprostol Kopen, Generieke misoprostol met paypal - Fiscalité pour tous | La lettre fiscale belge
Misoprostol Kopen, Generieke misoprostol met paypal - Fiscalité pour tous | La lettre fiscale belge Maladie de Tay-Sachs et maladie de Sandhoff - Pédiatrie - Édition professionnelle du Manuel MSD
Maladie de Tay-Sachs et maladie de Sandhoff - Pédiatrie - Édition professionnelle du Manuel MSD Transfert de gène in vivo chez la souris Sandhoff par des vecteurs (...) - Vaincre les Maladies Lysosomales
Transfert de gène in vivo chez la souris Sandhoff par des vecteurs (...) - Vaincre les Maladies Lysosomales Centre hospitalier universitaire à Limoges - CHU à Limoges
Centre hospitalier universitaire à Limoges - CHU à Limoges DeCS
DeCS