Maladie De Huntington
Expansion Triplets Répétés
Répétition Trinucléotide
Chromosomes Humains De La Paire 14
Protéines Tissu Nerveux
Acide Quinolinique
Protéines Nucléaires
Corps Strié
3-Hydroxyanthranilate 3,4-Dioxygenase
Age of Onset
Chorée
Rats, Transgenic
Maladies Expérimentales
Sorcellerie
Maladies Neurodégénératives
Genetic Testing
Souris Transgéniques
Test Du Rotarod
Atrophie
Encéphale
Striatum
Tétrabénazine
Mythologie
Acides Quinoliniques
Mutation
Peptides
Neurofibres Non-Myélinisées
Neurones
Corps D'Inclusion
Putamen
Phosphoprotéine Darpp-32 Régulée Par La Dopamine Et L'Ampc
Généalogie
Bande Chromosomique
Gene Knock-In Techniques
Marqueur Génétique
Phénotype
Allèles
Disease Progression
Conseil Génétique
Genetic Linkage
Nitrés, Composés
Diagnostic Précoce
Remnographie
Génotype
Troubles Cognitifs
Dégénérescence Nerveuse
Cartographie Chromosomique
Autophagie
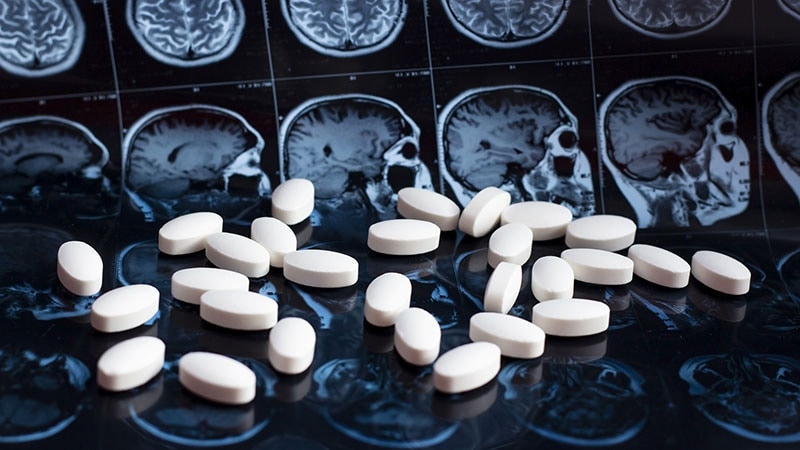
La maladie de Huntington est une maladie héréditaire et dégénérative du système nerveux. C'est un type de trouble neurologique qui affecte le mouvement, la cognition (pensée, raisonnement, apprentissage) et le comportement. La maladie est causée par une mutation dans un gène spécifique appelé HTT, qui code pour une protéine appelée huntingtine. Dans la maladie de Huntington, il y a une expansion anormale d'une séquence de répétition de nucléotides (un groupe de trois lettres de l'ADN) dans le gène HTT, ce qui entraîne la production d'une forme anormale et toxique de la protéine huntingtine.
Les symptômes de la maladie de Huntington commencent généralement entre 30 et 50 ans, bien qu'ils puissent apparaître à un âge plus précoce ou plus tardif. Les premiers signes comprennent souvent des mouvements corporels involontaires (chorée), de la difficulté à coordonner les mouvements, des changements d'humeur et de comportement, ainsi que des problèmes cognitifs subtils. Au fil du temps, les symptômes s'aggravent et peuvent inclure une détérioration mentale progressive, une perte de mémoire, des difficultés à parler et à avaler, une perte de poids et une diminution de la capacité à effectuer les activités quotidiennes.
Actuellement, il n'existe pas de traitement curatif pour la maladie de Huntington. Le traitement est axé sur la gestion des symptômes et l'amélioration de la qualité de vie des personnes atteintes. Les options de traitement peuvent inclure des médicaments pour contrôler les mouvements anormaux, des thérapies de réadaptation pour aider à maintenir les capacités fonctionnelles et un soutien psychologique pour faire face aux défis émotionnels et sociaux associés à la maladie.
Les expansions de triplets répétés (ETR) sont des mutations génétiques dans lesquelles s'observent des séquences d'ADN répétitives. Ces répétitions consistent généralement en une suite de trois nucléotides qui se copient plusieurs fois de manière consécutive. Chez les personnes atteintes d'ETR, ces répétitions sont anormalement longues et peuvent s'allonger encore plus au fil des générations, entraînant une variété de maladies neurodégénératives et autres troubles héréditaires.
Les ETR se produisent le plus souvent dans les régions non codantes de l'ADN, qui ne contiennent pas d'instructions pour la production de protéines. Cependant, lorsque ces répétitions se trouvent dans des gènes spécifiques, elles peuvent perturber la fonction génique et entraîner une maladie.
Les ETR sont associés à un large éventail de troubles héréditaires, notamment :
1. Maladie de Huntington - Une maladie neurodégénérative qui affecte le mouvement, la cognition et le comportement.
2. Ataxies spinocérébelleuses (SCA) - Un groupe de troubles du cervelet qui affectent l'équilibre, la coordination et les mouvements volontaires.
3. Myotonie de Steinert - Une maladie neuromusculaire caractérisée par une raideur musculaire et des crampes.
4. Maladie de la fragile X - Un trouble du développement qui peut causer des retards intellectuels, des problèmes d'élocution et des traits physiques distinctifs.
5. Dystrophie myotonique de type 1 (DM1) - Une maladie neuromusculaire caractérisée par une faiblesse musculaire progressive, une cataracte et des problèmes cardiaques.
Le nombre de répétitions d'une séquence nucléotidique spécifique dans un gène est directement lié à la gravité et à l'âge d'apparition de ces maladies. Plus le nombre de répétitions est élevé, plus les symptômes sont graves et plus ils apparaissent tôt dans la vie. Ce phénomène est connu sous le nom d'expansion de la répétition des trinucléotides.
Une répétition trinucléotide est un type spécifique de mutation génétique dans laquelle une séquence de trois nucléotides (les unités de base de l'ADN) est répétée de manière anormale et excessive dans une région donnée du gène. Normalement, ces répétitions sont présentes en petit nombre dans le génome, mais lorsqu'elles sont multipliées, elles peuvent entraîner des modifications de la structure et de la fonction des protéines codées par les gènes concernés.
Les répétitions trinucléotides sont souvent associées à certaines maladies neurodégénératives héréditaires, telles que la maladie de Huntington, la maladie de Charcot-Marie-Tooth et la sclérose latérale amyotrophique spinale (SLA) familiale. Le nombre de répétitions peut influencer l'âge d'apparition des symptômes et la sévérité de la maladie. Plus le nombre de répétitions est élevé, plus les symptômes sont susceptibles d'être précoces et graves.
Il est important de noter que les répétitions trinucléotides peuvent également être instables pendant la reproduction, ce qui signifie qu'elles ont tendance à s'allonger au fil des générations. Cela peut entraîner une anticipation génétique, où chaque nouvelle génération présente des symptômes plus précoces et plus sévères de la maladie.
En résumé, les répétitions trinucléotides sont des mutations génétiques anormales dans lesquelles une séquence de trois nucléotides est répétée de manière excessive dans un gène donné, ce qui peut entraîner des maladies neurodégénératives héréditaires.
Les chromosomes humains de la paire 14, également connus sous le nom de chromosomes 14, sont des structures composées de ADN et protéines qui contiennent des gènes et se trouvent dans le noyau de chaque cellule du corps. Les chromosomes 14 sont une paire de chromosomes homologues, ce qui signifie qu'ils ont la même taille, la même forme et contiennent des gènes similaires aux mêmes emplacements le long de la chromosome.
Chaque personne a 23 paires de chromosomes dans chaque cellule de leur corps, pour un total de 46 chromosomes. Les chromosomes 14 sont la 14ème paire de ces chromosomes, et ils sont numérotés de 14 à 14, ce qui signifie qu'ils sont présents deux fois dans chaque cellule du corps.
Les chromosomes 14 contiennent des centaines de gènes qui fournissent des instructions pour la production de protéines et d'autres produits génétiques importants pour le fonctionnement normal du corps. Les mutations dans ces gènes peuvent entraîner diverses maladies et conditions, telles que les troubles neurodégénératifs, les cancers et les maladies héréditaires.
En résumé, les chromosomes humains de la paire 14 sont des structures composées d'ADN et de protéines qui contiennent des gènes importants pour le fonctionnement normal du corps. Les mutations dans ces gènes peuvent entraîner diverses maladies et conditions.
Les protéines du tissu nerveux sont des types spécifiques de protéines qui se trouvent dans les neurones et le tissu nerveux périphérique. Elles jouent un rôle crucial dans la structure, la fonction et la régulation des cellules nerveuses. Parmi les protéines du tissu nerveux les plus importantes, on peut citer:
1. Neurofilaments: Ces protéines forment une partie importante de la structure interne des neurones et aident à maintenir leur intégrité structurelle. Elles sont également utilisées comme marqueurs pour diagnostiquer certaines maladies neurodégénératives.
2. Neurotransmetteurs: Ces protéines sont responsables de la transmission des signaux chimiques entre les neurones. Les exemples incluent la sérotonine, la dopamine et l'acétylcholine.
3. Canaux ioniques: Ces protéines régulent le flux d'ions à travers la membrane cellulaire des neurones, ce qui est essentiel pour la génération et la transmission des impulsions nerveuses.
4. Protéines d'adhésion: Elles aident à maintenir les contacts entre les neurones et d'autres types de cellules dans le tissu nerveux.
5. Enzymes: Les protéines enzymatiques sont importantes pour la régulation des processus métaboliques dans les neurones, y compris la synthèse et la dégradation des neurotransmetteurs.
6. Chaperons moléculaires: Ces protéines aident à plier et à assembler d'autres protéines dans les neurones, ce qui est essentiel pour leur fonction et leur survie.
7. Protéines de structure: Elles fournissent une structure et un soutien aux cellules nerveuses, telles que la tubuline, qui forme des microtubules dans le cytosquelette des neurones.
Des anomalies dans les protéines du tissu nerveux peuvent entraîner divers troubles neurologiques, y compris des maladies neurodégénératives telles que la maladie d'Alzheimer et la maladie de Parkinson.
L'acide quinolique est un métabolite endogène que l'on trouve normalement dans le cerveau et d'autres tissus du corps. Il est produit à partir de tryptophane, un acide aminé essentiel, par une série de réactions enzymatiques.
Dans des conditions normales, les niveaux d'acide quinolique dans le cerveau sont régulés et maintenus à des niveaux bas. Cependant, dans certaines conditions pathologiques, telles que l'inflammation, l'ischémie ou l'infection, la production d'acide quinolique peut être augmentée, entraînant une accumulation toxique de ce métabolite dans le cerveau.
Des niveaux élevés d'acide quinolique ont été impliqués dans plusieurs processus pathologiques du cerveau, notamment la neuroinflammation, l'excitotoxicité et la mort cellulaire neuronale. En particulier, il a été suggéré que l'acide quinolique pourrait jouer un rôle important dans le développement et la progression de maladies neurodégénératives telles que la sclérose en plaques, la maladie de Parkinson et la maladie d'Alzheimer.
En outre, des études ont montré que l'acide quinolique peut potentialiser les effets toxiques de certaines protéines mal repliées associées à ces maladies neurodégénératives. Par conséquent, il est considéré comme une cible thérapeutique potentielle pour le traitement de ces maladies.
En résumé, l'acide quinolique est un métabolite endogène qui peut avoir des effets toxiques sur le cerveau à des niveaux élevés. Il a été impliqué dans plusieurs processus pathologiques du cerveau et est considéré comme une cible thérapeutique potentielle pour le traitement de maladies neurodégénératives.
Les protéines nucléaires sont des protéines qui se trouvent dans le noyau des cellules et jouent un rôle crucial dans la régulation de l'expression des gènes, la réplication de l'ADN, la réparation de l'ADN, la transcription de l'ARN et d'autres processus essentiels à la survie et à la reproduction des cellules.
Il existe plusieurs types de protéines nucléaires, y compris les histones, qui sont des protéines structurelles qui aident à compacter l'ADN en chromosomes, et les facteurs de transcription, qui se lient à l'ADN pour réguler l'expression des gènes. Les protéines nucléaires peuvent également inclure des enzymes qui sont impliquées dans la réplication et la réparation de l'ADN, ainsi que des protéines qui aident à maintenir l'intégrité structurelle du noyau.
Les protéines nucléaires peuvent être régulées au niveau de leur expression, de leur localisation dans la cellule et de leur activité enzymatique. Des anomalies dans les protéines nucléaires peuvent entraîner des maladies génétiques et contribuer au développement du cancer. Par conséquent, l'étude des protéines nucléaires est importante pour comprendre les mécanismes moléculaires qui sous-tendent la régulation de l'expression des gènes et d'autres processus cellulaires essentiels.
Le corps strié, également connu sous le nom de striatum, est une structure importante du cerveau qui fait partie du système nerveux central. Il s'agit d'une région complexe composée de deux parties principales : le putamen et le noyau caudé, ainsi que d'une petite zone appelée le globus pallidus interne.
Le corps strié joue un rôle crucial dans la régulation du mouvement volontaire et involontaire, ainsi que dans les processus de récompense et de motivation. Il reçoit des informations des noyaux gris centraux et des cortex sensorimoteurs, et envoie des signaux aux noyaux gris centraux et au thalamus.
Les neurones du corps strié utilisent principalement le neurotransmetteur dopamine pour communiquer avec d'autres cellules nerveuses. Les déséquilibres dans la transmission de la dopamine peuvent entraîner des troubles du mouvement, tels que la maladie de Parkinson et la chorée de Huntington.
En plus de son rôle dans le mouvement, le corps strié est également impliqué dans les processus cognitifs supérieurs, tels que l'apprentissage et la mémoire, ainsi que dans les émotions et les comportements liés à la récompense. Des recherches récentes ont également suggéré qu'il pourrait jouer un rôle dans la régulation de l'humeur et des fonctions exécutives.
La 3-Hydroxyanthranilate 3,4-Dioxygenase est une enzyme qui joue un rôle important dans la dégradation de l'acide tryptophane dans notre corps. Cette enzyme catalyse la réaction chimique qui oxygène le 3-hydroxyanthranilate pour former 2-amino-3-carboxymuconate semi-aldehyde, un intermédiaire important dans le métabolisme de l'acide tryptophane. Ce processus est régulé par plusieurs facteurs et est associé à diverses voies métaboliques et physiologiques, y compris la réponse immunitaire et la production d'acides biliaires. Des anomalies dans le fonctionnement de cette enzyme peuvent être liées à certaines maladies, telles que la maladie de Parkinson et la porphyrie.
L'âge d'apparition, également connu sous le nom d'âge de début ou d'âge de survenue, fait référence à l'âge auquel une personne développe pour la première fois les symptômes ou manifestations d'une maladie, d'un trouble de santé mentale, d'un handicap ou d'autres conditions médicales. Il peut être exprimé en années, mois ou même semaines après la naissance, selon le type et la gravité de la condition concernée.
L'âge d'apparition est un aspect important du diagnostic et de la prise en charge médicale, car il peut fournir des indices sur les causes sous-jacentes de la maladie, influencer le choix des traitements et des interventions, et aider à prévoir l'évolution et le pronostic de la condition.
Par exemple, certaines maladies génétiques ou congénitales peuvent se manifester dès la naissance ou dans les premiers mois de vie, tandis que d'autres troubles, tels que la maladie d'Alzheimer ou la démence, ne se développent généralement qu'à un âge plus avancé. De même, certains troubles mentaux, comme l'autisme et le trouble déficitaire de l'attention avec hyperactivité (TDAH), peuvent présenter des signes avant-coureurs dès la petite enfance, tandis que d'autres, comme la schizophrénie, peuvent ne se manifester qu'à l'adolescence ou à l'âge adulte.
Il est important de noter que l'âge d'apparition peut varier considérablement d'une personne à l'autre, même au sein d'une même famille ou d'un même groupe de population. Des facteurs tels que les antécédents familiaux, l'environnement, le mode de vie et d'autres facteurs de risque peuvent influencer le moment où une personne développe les symptômes d'une condition donnée.
La chorée est un trouble du mouvement caractérisé par des mouvements involontaires, irréguliers et souvent rapides des muscles du visage, du tronc et des membres. Ces mouvements peuvent être continus ou se produire de manière intermittente. La chorée est généralement observée dans certaines maladies neurologiques, telles que la maladie de Huntington, la chorée de Sydenham (une complication du rhumatisme articulaire aigu), les troubles hépatiques et certains types d'intoxications. Dans certains cas, la chorée peut être un effet secondaire de certains médicaments, comme les antipsychotiques.
La cause sous-jacente de la chorée est généralement une anomalie dans le fonctionnement des neurones qui produisent et régulent la dopamine, un neurotransmetteur important dans le cerveau. Selon la cause sous-jacente, la chorée peut être traitée en traitant la maladie sous-jacente ou en ajustant les médicaments qui peuvent causer des mouvements involontaires. Dans certains cas, des médicaments spécifiques peuvent être utilisés pour contrôler les symptômes de la chorée.
Les symptômes prodromiques sont des signes ou des symptômes avant-coureurs qui peuvent indiquer qu'une personne va développer une maladie, un trouble ou une crise particulière. Ces symptômes peuvent être physiques, cognitifs ou émotionnels et peuvent varier en fonction de la condition sous-jacente.
Dans certains cas, les symptômes prodromiques peuvent être subtils et facilement ignorés ou attribués à d'autres causes. Cependant, ils peuvent également être assez graves pour affecter considérablement la fonction quotidienne d'une personne.
Par exemple, dans la schizophrénie, les symptômes prodromiques peuvent inclure des changements de comportement, des difficultés de concentration, une diminution de la motivation et des expériences sensorielles inhabituelles. Dans l'épilepsie, les symptômes prodromiques peuvent inclure des maux de tête, des nausées, des étourdissements ou des changements d'humeur avant une crise.
Il est important de noter que la présence de symptômes prodromiques ne signifie pas nécessairement qu'une personne développera définitivement une condition donnée. Cependant, si quelqu'un remarque des symptômes prodromiques persistants ou inexpliqués, il est recommandé de consulter un professionnel de la santé pour une évaluation et des soins appropriés.
Les rats transgéniques sont des rongeurs qui ont été génétiquement modifiés en insérant des gènes étrangers dans leur génome. Cela permet aux chercheurs d'étudier les effets de ces gènes sur le développement, la physiologie et le comportement des rats. Les gènes peuvent être prélevés sur d'autres espèces, tels que les humains, ou ils peuvent être synthétisés en laboratoire.
L'insertion de ces gènes étrangers dans le génome du rat est souvent réalisée en utilisant des vecteurs viraux, tels que les rétrovirus ou les lentivirus, qui sont capables d'intégrer leur matériel génétique dans celui de l'hôte. Les gènes étrangers peuvent également être délivrés directement dans l'embryon à l'aide de techniques d'injection microscopiques.
Les rats transgéniques sont largement utilisés dans la recherche biomédicale pour étudier diverses maladies, telles que le cancer, les maladies cardiovasculaires, le diabète et les maladies neurodégénératives. Ils offrent une alternative avantageuse aux modèles murins traditionnels en raison de leur taille plus grande, ce qui facilite la réalisation d'interventions chirurgicales et la collecte de données physiologiques. De plus, les rats sont génétiquement plus proches des humains que les souris, ce qui signifie qu'ils peuvent être utilisés pour étudier des maladies complexes qui ne se manifestent pas chez les souris.
Cependant, il est important de noter que les rats transgéniques sont des organismes génétiquement modifiés et qu'il existe des préoccupations éthiques associées à leur utilisation dans la recherche. Il est donc essentiel de veiller à ce que leur utilisation soit réglementée et encadrée par des protocoles stricts pour garantir le bien-être des animaux et la validité scientifique des résultats obtenus.
Je suis désolé, mais la phrase "Maladies Expérimentales" ne correspond pas à une définition médicale spécifique. Le terme "maladie expérimentale" peut faire référence à des études ou recherches scientifiques dans lesquelles des maladies sont simulées ou créées dans un laboratoire, généralement sur des animaux de laboratoire, pour mieux comprendre la pathogenèse, le développement et les traitements possibles d'une maladie. Cependant, il ne s'agit pas d'un terme médical standardisé. Si vous cherchez des informations sur une procédure ou un concept médical spécifique, veuillez me fournir plus de détails afin que je puisse vous aider au mieux.
La sorcellerie ne fait pas partie des termes reconnus dans la médecine ou la science médicale. Il s'agit plutôt d'une croyance culturelle ou religieuse selon laquelle certaines personnes peuvent utiliser des forces surnaturelles ou magiques pour infliger du mal ou causer des événements indésirables. Ces croyances varient considérablement parmi différentes cultures et sociétés, et elles ne sont pas soutenues par la preuve scientifique. Dans un contexte médical, on s'efforcerait de comprendre les causes sous-jacentes des symptômes ou des préoccupations d'une personne en termes de processus physiologiques et mentaux établis, plutôt que d'attribuer des événements à la sorcellerie.
Les maladies neurodégénératives sont un groupe de conditions médicales progressives qui impliquent une dégénérescence et une mort cellulaire dans les neurones ou les cellules nerveuses du cerveau. Ce processus entraîne des dommages aux connections nerveuses et, finalement, à la perte des fonctions neurologiques essentielles.
Les symptômes de ces maladies varient en fonction de la région du cerveau qui est affectée. Ils peuvent inclure des problèmes cognitifs comme la démence, des problèmes moteurs tels que les tremblements ou la difficulté à bouger, et d'autres problèmes comme la perte de l'odorat ou la dépression.
Les exemples courants de maladies neurodégénératives comprennent la maladie d'Alzheimer, la maladie de Parkinson, la sclérose latérale amyotrophique (SLA), la démence à corps de Lewy, et la maladie de Huntington. Ces maladies sont généralement incurables et les traitements disponibles visent principalement à soulager les symptômes et à améliorer la qualité de vie des patients.
Le noyau caudé est une structure importante du système nerveux central, plus spécifiquement dans le diencéphale. Il fait partie du striatum, qui est un élément clé du circuit de la boucle cortico-striato-thalamo-corticale. Cette boucle joue un rôle crucial dans la planification et l'exécution des mouvements volontaires, ainsi que dans les processus cognitifs et émotionnels.
Le noyau caudé a une forme allongée et est divisé en deux parties : le noyau caudé dorsal et le noyau caudé ventral. Il contient principalement des neurones GABAergiques (qui libèrent de l'acide gamma-aminobutyrique) et des neurones à cholinergiques.
Les afférences au noyau caudé proviennent principalement du cortex cérébral, tandis que ses efférences se dirigent vers le globus pallidus interne (GPi), le globus pallidus externe (GPe) et le thalamus. Les connexions avec ces structures sont essentielles pour la régulation des mouvements volontaires et involontaires, ainsi que pour les processus d'apprentissage et de mémoire.
Des anomalies dans la structure ou la fonction du noyau caudé ont été associées à divers troubles neurologiques et psychiatriques, tels que la maladie de Parkinson, la chorée de Huntington, les troubles obsessionnels compulsifs (TOC) et la schizophrénie.
Le test génétique est un type d'examen diagnostique qui consiste à analyser les gènes d'un individu dans le but d'identifier des modifications ou des variations dans l'ADN qui peuvent être associées à un risque accru de développer une maladie héréditaire ou d'autres conditions médicales. Les tests génétiques peuvent également être utilisés pour déterminer la susceptibilité d'un individu à répondre à certains traitements médicaux ou pour établir des relations familiales.
Les tests génétiques peuvent impliquer l'analyse de l'ADN, de l'ARN ou des protéines, et peuvent être effectués sur des échantillons de sang, de salive, de cheveux ou d'autres tissus. Les résultats du test génétique peuvent aider les médecins à poser un diagnostic, à prévoir le risque de développer une maladie à l'avenir, à déterminer le meilleur traitement pour une maladie existante ou à fournir des conseils en matière de planification familiale.
Il est important de noter que les tests génétiques peuvent avoir des implications émotionnelles et sociales importantes pour les individus et leur famille, et qu'il est donc essentiel de recevoir un counseling génétique avant et après le test pour comprendre pleinement les résultats et les conséquences potentielles.
Les souris transgéniques sont un type de souris génétiquement modifiées qui portent et expriment des gènes étrangers ou des séquences d'ADN dans leur génome. Ce processus est accompli en insérant le gène étranger dans l'embryon précoce de la souris, généralement au stade une cellule, ce qui permet à la modification de se propager à toutes les cellules de l'organisme en développement.
Les souris transgéniques sont largement utilisées dans la recherche biomédicale pour étudier la fonction et le rôle des gènes spécifiques dans le développement, la physiologie et la maladie. Elles peuvent être utilisées pour modéliser diverses affections humaines, y compris les maladies génétiques, le cancer, les maladies cardiovasculaires et neurologiques.
Les chercheurs peuvent concevoir des souris transgéniques avec des caractéristiques spécifiques en insérant un gène particulier qui code pour une protéine d'intérêt ou en régulant l'expression d'un gène endogène. Cela permet aux chercheurs de mieux comprendre les voies moléculaires et cellulaires impliquées dans divers processus physiologiques et pathologiques, ce qui peut conduire à de nouvelles stratégies thérapeutiques pour traiter les maladies humaines.
Le test du rotarod est un communément utilisé dans les études précliniques pour évaluer l'équilibre, la coordination et la force musculaire des animaux, comme les souris et les rats. Dans ce test, l'animal est placé sur une barre rotative horizontale qui tourne à différentes vitesses. L'animal doit alors maintenir son équilibre et marcher sur la barre aussi longtemps que possible sans tomber.
Les chercheurs peuvent mesurer le temps qu'un animal est capable de rester sur la barre rotative, ce qui leur permet d'évaluer l'effet de différents traitements ou interventions sur l'équilibre et la coordination de l'animal. Ce test est souvent utilisé pour évaluer les effets des médicaments ou des interventions qui peuvent affecter le système nerveux central, comme les agents neuroprotecteurs, les analgésiques ou les agents qui affectent la fonction musculaire.
Le test du rotarod est considéré comme une méthode fiable et sensible pour évaluer l'équilibre et la coordination des animaux de laboratoire. Il est largement utilisé dans la recherche fondamentale et translationnelle, y compris dans le développement de nouveaux médicaments et thérapies pour les troubles neurologiques et musculaires.
L'atrophie est un terme médical qui décrit la diminution de la taille ou du volume d'un tissu, d'un organe ou d'une partie du corps en raison de la perte de cellules ou de la réduction de leur taille. Cela peut être causé par une variété de facteurs, y compris le vieillissement, les maladies chroniques, l'inactivité physique, la dénutrition et les lésions nerveuses.
Les exemples courants d'atrophie comprennent la fonte musculaire due à l'immobilisation prolongée, la perte de tissu cérébral dans des conditions telles que la maladie d'Alzheimer ou la sclérose en plaques, et la réduction de la taille de la glande mammaire chez les femmes qui allaitent.
Les symptômes de l'atrophie dépendent de la zone du corps affectée. Ils peuvent inclure une faiblesse musculaire, une perte d'équilibre, des mouvements plus lents et moins précis, une diminution de la fonction sensorielle, une modification de la voix ou de la vision, et dans certains cas, des douleurs ou des crampes.
Le traitement de l'atrophie dépend de la cause sous-jacente. Dans certains cas, il peut être possible de ralentir ou d'arrêter le processus d'atrophie en traitant la maladie sous-jacente. Dans d'autres cas, des exercices de renforcement musculaire, une thérapie physique ou occupationnelle, et des changements de mode de vie peuvent aider à améliorer les symptômes et la fonction.
L'encéphale est la structure centrale du système nerveux situé dans la boîte crânienne. Il comprend le cerveau, le cervelet et le tronc cérébral. L'encéphale est responsable de la régulation des fonctions vitales telles que la respiration, la circulation sanguine et la température corporelle, ainsi que des fonctions supérieures telles que la pensée, la mémoire, l'émotion, le langage et la motricité volontaire. Il est protégé par les os de la boîte crânienne et recouvert de trois membranes appelées méninges. Le cerveau et le cervelet sont floating dans le liquide céphalo-rachidien, qui agit comme un coussin pour amortir les chocs et les mouvements brusques.
Le striatum est une structure importante du cerveau qui fait partie du système nerveux central. Il s'agit d'une région du mésencéphale située dans le noyau gris central et constitue la principale entrée du système nerveux extrapyramidal, qui est responsable du contrôle moteur involontaire et de l'apprentissage procédural.
Le striatum est divisé en deux parties principales : le noyau caudé et le putamen, qui sont souvent considérés ensemble sous le nom de néostriatum, et le globus pallidus, qui est divisé en deux segments, le globus pallidus interne (GPi) et le globus pallidus externe (GPe).
Le striatum reçoit des afférences importantes du cortex cérébral et de la substance noire, et envoie des efférences au globus pallidus et au nucléus subthalamicus. Il joue un rôle crucial dans la régulation de la motricité, de l'apprentissage, de la récompense, de l'addiction et de certaines fonctions cognitives.
Des anomalies du striatum ont été impliquées dans divers troubles neurologiques et psychiatriques, tels que la maladie de Parkinson, la chorée de Huntington, les troubles obsessionnels compulsifs (TOC) et la schizophrénie.
La tétrabénazine est un médicament prescrit pour le traitement des symptômes moteurs associés à la maladie de Huntington, une maladie neurologique héréditaire. Il agit en réduisant la quantité de dopamine, un neurotransmetteur, dans le cerveau. La tétrabénazine se lie aux vésicules qui stockent la dopamine et provoque leur fusion, entraînant une diminution des niveaux de dopamine disponibles pour activer les récepteurs postsynaptiques. Cela peut aider à contrôler les mouvements involontaires excessifs (chorée) caractéristiques de la maladie de Huntington.
Les effets secondaires courants de la tétrabénazine comprennent la somnolence, la fatigue, la nausée, la constipation et les étourdissements. Les effets secondaires plus graves peuvent inclure des problèmes psychiatriques tels que la dépression, l'anxiété, l'agitation ou la pensée suicidaire, ainsi qu'une augmentation du risque de syndrome neuroleptique malin (SNM), une affection potentiellement mortelle qui affecte le système nerveux. Par conséquent, la tétrabénazine doit être utilisée avec prudence et sous surveillance médicale étroite.
Il est important de noter que la tétrabénazine n'est pas une cure pour la maladie de Huntington et qu'elle ne peut pas ralentir ou arrêter sa progression. Il vise uniquement à soulager les symptômes moteurs associés à cette maladie.
Je suis désolé, mais il semble y avoir une certaine confusion dans votre question. La mythologie ne fait pas partie des termes médicaux. La mythologie est l'étude des mythes, qui sont souvent des histoires symboliques ou métaphoriques utilisées pour expliquer des aspects du monde naturel ou de la société, des croyances religieuses, des concepts philosophiques, des événements historiques ou des instructions morales dans une culture donnée. Ils peuvent inclure des dieux, des déesses, des héros et d'autres êtres surnaturels, ainsi que leurs aventures.
Si vous cherchez une définition médicale, je suis heureux de vous aider avec cela. Cependant, je dois noter qu'il est important de poser des questions claires et précises pour recevoir les réponses les plus utiles et exactes possibles.
Les acides quinoléiques sont des métabolites endogènes qui sont produits dans le corps humain lors du métabolisme de tryptophane, un acide aminé essentiel. Il existe deux principaux types d'acides quinoléiques : l'acide 3-hydroxykynurénine (3HK) et l'acide quinolinique (AQ).
Dans des conditions normales, les acides quinoléiques sont présents à des niveaux relativement faibles dans le corps. Mais dans certaines situations, telles que l'inflammation chronique, l'infection, le stress oxydatif ou la dégradation accrue du tryptophane, les niveaux d'acides quinoléiques peuvent augmenter considérablement.
Les acides quinoléiques ont des effets complexes sur le corps humain. D'une part, ils sont nécessaires pour la synthèse de la nicotinamide adénine dinucléotide (NAD+), une molécule essentielle au métabolisme énergétique et à d'autres processus cellulaires importants.
Cependant, des niveaux excessifs d'acides quinoléiques, en particulier l'acide quinolinique, peuvent être toxiques pour le cerveau et ont été associés à un certain nombre de maladies neurologiques, notamment la maladie de Parkinson, la sclérose en plaques et la maladie d'Alzheimer.
L'acide quinolinique est également un ligand du récepteur N-méthyl-D-aspartate (NMDA) du glutamate, qui joue un rôle important dans la transmission des signaux nerveux dans le cerveau. Des niveaux élevés d'acide quinolinique peuvent entraîner une activation excessive des récepteurs NMDA, ce qui peut endommager les neurones et contribuer au développement de maladies neurodégénératives.
En résumé, l'acide quinolinique est un métabolite important du tryptophane qui joue un rôle crucial dans la production de NAD+ et la transmission des signaux nerveux dans le cerveau. Cependant, des niveaux excessifs d'acide quinolinique peuvent être toxiques pour le cerveau et ont été associés à un certain nombre de maladies neurologiques. Par conséquent, il est important de maintenir des niveaux équilibrés d'acide quinolinique dans le corps pour prévenir les dommages neuronaux et protéger la santé du cerveau.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
Les peptides sont de courtes chaînes d'acides aminés, liés entre eux par des liaisons peptidiques. Ils peuvent contenir jusqu'à environ 50 acides aminés. Les peptides sont produits naturellement dans le corps humain et jouent un rôle crucial dans de nombreuses fonctions biologiques, y compris la signalisation cellulaire et la régulation hormonale. Ils peuvent également être synthétisés en laboratoire pour une utilisation dans la recherche médicale et pharmaceutique. Les peptides sont souvent utilisés comme médicaments car ils peuvent se lier sélectivement à des récepteurs spécifiques et moduler leur activité, ce qui peut entraîner une variété d'effets thérapeutiques.
Il existe de nombreux types différents de peptides, chacun ayant des propriétés et des fonctions uniques. Certains peptides sont des hormones, comme l'insuline et l'hormone de croissance, tandis que d'autres ont des effets anti-inflammatoires ou antimicrobiens. Les peptides peuvent également être utilisés pour traiter une variété de conditions médicales, telles que la douleur, l'arthrite, les maladies cardiovasculaires et le cancer.
Dans l'ensemble, les peptides sont des molécules importantes qui jouent un rôle clé dans de nombreux processus biologiques et ont des applications prometteuses dans le domaine médical et pharmaceutique.
Les neurofibres non-myélinisées sont des fibres nerveuses dans le système nerveux périphérique qui ne sont pas entourées d'une gaine de myéline. La myéline est une substance grasse produite par les cellules gliales, qui agit comme un isolant électrique pour accélérer la conduction des impulsions nerveuses.
Dans le cas des neurofibres non-myélinisées, les axones (les prolongements des neurones) sont directement en contact avec l'environnement extracellulaire et sont donc exposés aux interférences extérieures. Ces fibres nerveuses sont généralement plus petites que les neurofibres myélinisées et conduisent les impulsions nerveuses plus lentement.
Les neurofibres non-myélinisées jouent un rôle important dans la transmission de signaux sensoriels tels que la douleur, la température et le toucher léger. Les dommages ou les dysfonctionnements de ces fibres peuvent entraîner des troubles neurologiques tels que la neuropathie périphérique, qui peut se manifester par des symptômes tels que des douleurs, des picotements, des engourdissements et une faiblesse musculaire.
Les neurones, également connus sous le nom de cellules nerveuses, sont les unités fonctionnelles fondamentales du système nerveux. Ils sont responsables de la réception, du traitement, de la transmission et de la transduction des informations dans le cerveau et d'autres parties du corps. Les neurones se composent de trois parties principales : le dendrite, le corps cellulaire (ou soma) et l'axone.
1. Les dendrites sont des prolongements ramifiés qui reçoivent les signaux entrants d'autres neurones ou cellules sensoriques.
2. Le corps cellulaire contient le noyau de la cellule, où se trouvent l'ADN et les principales fonctions métaboliques du neurone.
3. L'axone est un prolongement unique qui peut atteindre une longueur considérable et transmet des signaux électriques (potentiels d'action) vers d'autres neurones ou cellules effectrices, telles que les muscles ou les glandes.
Les synapses sont les sites de communication entre les neurones, où l'axone d'un neurone se connecte aux dendrites ou au corps cellulaire d'un autre neurone. Les neurotransmetteurs sont des molécules chimiques libérées par les neurones pour transmettre des signaux à travers la synapse vers d'autres neurones.
Les neurones peuvent être classés en différents types en fonction de leur morphologie, de leurs propriétés électriques et de leur rôle dans le système nerveux. Par exemple :
- Les neurones sensoriels capturent et transmettent des informations sensorielles provenant de l'environnement externe ou interne vers le cerveau.
- Les neurones moteurs transmettent les signaux du cerveau vers les muscles ou les glandes pour provoquer une réponse motrice ou hormonale.
- Les interneurones sont des neurones locaux qui assurent la communication et l'intégration entre les neurones sensoriels et moteurs dans le système nerveux central.
Un corps d'inclusion est un terme utilisé en histopathologie et en biologie cellulaire pour décrire une inclusion cytoplasmique anormale dans les cellules. Ces inclusions sont généralement constituées de matériel anormal accumulé dans la cellule, comme des protéines mal repliées ou des granules lipidiques. Les corps d'inclusion peuvent être observés dans diverses affections, y compris certaines maladies neurodégénératives et infections virales.
Les corps d'inclusion peuvent varier en taille, en forme et en apparence selon leur composition et la maladie associée. Certains sont sphériques ou ovoïdes, tandis que d'autres ont des formes plus complexes. Ils peuvent être hyalins, granulaires, vasculaires ou cristallins. Les corps d'inclusion peuvent contenir des protéines anormales telles que l'alpha-synucléine dans la maladie de Parkinson et la protéine tau dans la maladie d'Alzheimer.
Les mécanismes sous-jacents à la formation des corps d'inclusion ne sont pas entièrement compris, mais on pense qu'ils résultent d'un dysfonctionnement du système de contrôle de la qualité des protéines ou d'une réponse anormale au stress cellulaire. Les corps d'inclusion peuvent contribuer aux dommages et à la mort des cellules, ce qui entraîne une perte de fonction tissulaire et peut conduire à des maladies graves.
Il est important de noter que tous les corps d'inclusion ne sont pas pathologiques et peuvent être observés dans des cellules normales sous certaines conditions, comme le vieillissement ou l'exposition à des toxines environnementales. Cependant, la présence de corps d'inclusion dans certains contextes peut indiquer une maladie spécifique et être utile pour le diagnostic et la recherche sur les maladies neurodégénératives.
Le putamen est une structure cérébrale faisant partie du striatum, qui est un composant essentiel du système nerveux central. Il est situé dans le sous-cortex cérébral et fait partie des noyaux gris centraux. Le putamen a une forme de poire et est adjacent au pallidum, avec lequel il forme le néostriatum.
Le putamen joue un rôle crucial dans le contrôle moteur et les mouvements involontaires. Il participe également à l'apprentissage procédural, la motivation, la récompense et les processus cognitifs tels que la planification et la prise de décision. Le putamen reçoit des afférences importantes du cortex cérébral et projette vers le pallidum et d'autres structures sous-corticales, y compris la substance noire et le thalamus. Ces connexions sont essentielles aux circuits neuronaux qui sous-tendent les fonctions mentionnées ci-dessus.
Des anomalies du putamen ont été associées à divers troubles neurologiques et psychiatriques, tels que la maladie de Parkinson, la dystonie, la chorée de Huntington, la toxicomanie et les troubles obsessionnels compulsifs.
La phosphoprotéine Darpp-32 (Dopamine and cAMP-regulated Phosphoprotein of Mr 32 kDa) est une protéine régulatrice importante dans les neurones du cerveau. Elle joue un rôle crucial dans la modulation des signaux de neurotransmetteurs, en particulier ceux impliquant la dopamine et l'AMPc (adénylate cyclase activée par le récepteur de l'adrénaline).
Darpp-32 est rapidement phosphorylée et déphosphorylée en réponse aux changements dans les niveaux de dopamine et d'AMPc. Cette modification post-traductionnelle permet à Darpp-32 de réguler l'activité des protéines kinases et des protéines phosphatases, ce qui a un impact sur la transmission synaptique et la plasticité neuronale.
La protéine Darpp-32 est fortement exprimée dans les neurones du striatum, une région cérébrale clé pour le contrôle moteur et l'apprentissage associatif récompensé. Les déséquilibres de la régulation de Darpp-32 ont été impliqués dans plusieurs troubles neurologiques, tels que la maladie de Parkinson, les troubles obsessionnels compulsifs et la toxicomanie.
Les protéines mutantes sont des protéines dont la séquence d'acides aminés ou la structure tridimensionnelle est altérée en raison d'une mutation dans le gène qui les code. Les mutations peuvent être héréditaires ou spontanées et peuvent entraîner des changements dans la fonction, l'activité, la stabilité ou l'interaction de la protéine avec d'autres molécules. Ces modifications peuvent avoir des effets bénins, nuls ou graves sur la physiologie et la santé de l'organisme, en fonction de la protéine concernée et de la nature de la mutation. Certaines mutations de protéines peuvent être à l'origine de maladies génétiques, de maladies dégénératives ou de processus tumoraux.
En termes médicaux, la généalogie est l'étude systématique des antécédents familiaux et médicaux d'une personne ou d'une famille sur plusieurs générations. Elle vise à identifier les modèles de maladies héréditaires ou génétiques dans une famille, ce qui peut aider à évaluer le risque de développer certaines affections et à mettre en œuvre des stratégies de prévention et de dépistage appropriées.
Les professionnels de la santé utilisent souvent des arbres généalogiques pour représenter visuellement les relations familiales et les antécédents médicaux. Ces outils peuvent être particulièrement utiles dans la pratique clinique, en particulier lorsqu'il s'agit de maladies rares ou complexes qui ont tendance à se produire dans certaines familles en raison de facteurs génétiques sous-jacents.
En plus d'être un outil important pour la médecine préventive, la généalogie peut également fournir des informations précieuses sur l'histoire naturelle de diverses maladies et conditions, ce qui contribue à faire progresser notre compréhension globale de la pathogenèse et de la physiopathologie. Par conséquent, elle joue un rôle crucial dans la recherche médicale et les soins cliniques.
Une bande chromosomique est une section ou une région spécifique d'un chromosome qui a été identifiée et caractérisée par sa position, sa taille et son motif de bandes sombres et pâles lorsqu'il est visualisé au microscope à l'aide de techniques de coloration spéciales.
Les bandes chromosomiques sont un outil important en cytogénétique, qui est la branche de la génétique qui étudie les chromosomes et leur rôle dans l'hérédité et les maladies humaines. Les motifs de bandes caractéristiques permettent aux chercheurs d'identifier des anomalies chromosomiques spécifiques, telles que des délétions, des duplications ou des translocations, qui peuvent être associées à des troubles génétiques ou à des maladies.
Les bandes chromosomiques sont généralement nommées en fonction de leur position relative sur le bras court (p) ou le bras long (q) d'un chromosome particulier. Par exemple, la bande 12p13.1 est située dans la région 13.1 du bras court du chromosome 12. Les bandes sont également classées en fonction de leur largeur relative, allant des plus larges (bandes 1 à 5) aux plus étroites (bandes 6 à 10).
En résumé, les bandes chromosomiques sont des marqueurs visuels importants utilisés pour identifier et caractériser les anomalies chromosomiques associées à divers troubles génétiques et maladies.
La lipoylation est un processus biochimique dans lequel un résidu d'acide lipoïque, un cofacteur essentiel, est attaché à une protéine spécifique. L'acide lipoïque joue un rôle crucial dans les réactions redox dans le métabolisme énergétique, en particulier dans le cycle de Krebs, où il agit comme un transporteur d'électrons réversible. La lipoylation est une modification post-traductionnelle des protéines qui se produit dans le réticulum endoplasmique et les mitochondries. Ce processus est essentiel pour maintenir l'intégrité et la fonctionnalité du métabolisme énergétique et d'autres voies métaboliques importantes dans la cellule. Des anomalies dans le processus de lipoylation peuvent entraîner divers troubles métaboliques et maladies.
Les "Gene Knock-In Techniques" sont des méthodes utilisées en génie génétique pour modifier le génome d'un organisme en insérant un gène spécifique à un emplacement précis. Contrairement aux techniques de knockout où un gène est désactivé ou supprimé, le but du knock-in est d'introduire une mutation spécifique dans un gène existant ou d'insérer un gène étranger dans un locus chromosomique spécifique.
Ces techniques comprennent souvent l'utilisation de vecteurs viraux, tels que des adénovirus ou des lentivirus, pour délivrer le gène d'intérêt dans les cellules cibles. Une autre méthode courante consiste à utiliser des systèmes de recombinaison homologue pour insérer le gène au bon endroit dans le génome.
Les techniques de knock-in sont largement utilisées en recherche biomédicale pour étudier les fonctions des gènes, ainsi que pour développer des modèles animaux de maladies humaines. Elles ont également des applications potentielles dans le domaine de la thérapie génique, où elles peuvent être utilisées pour corriger des mutations génétiques spécifiques associées à des maladies héréditaires.
Un marqueur génétique est un segment spécifique de l'ADN qui est variable d'une personne à l'autre. Il peut être utilisé pour identifier des individus ou des groupes d'individus partageant des caractéristiques particulières, comme une prédisposition à certaines maladies. Les marqueurs génétiques ne causent pas directement la maladie, mais ils peuvent indiquer une région du génome où un gène associé à la maladie peut être situé.
Les marqueurs génétiques sont souvent utilisés dans la recherche médicale et la médecine prédictive pour aider à diagnostiquer des conditions héréditaires, prédire le risque de développer certaines maladies, suivre l'évolution d'une maladie ou déterminer la réponse à un traitement spécifique. Ils peuvent également être utiles dans les enquêtes médico-légales pour identifier des victimes ou des auteurs de crimes.
Les marqueurs génétiques peuvent prendre différentes formes, telles que des variations dans la longueur des séquences répétitives d'ADN (VNTR), des polymorphismes nucléotidiques simples (SNP) ou des insertions/délétions de quelques paires de bases.
Le phénotype est le résultat observable de l'expression des gènes en interaction avec l'environnement et d'autres facteurs. Il s'agit essentiellement des manifestations physiques, biochimiques ou développementales d'un génotype particulier.
Dans un contexte médical, le phénotype peut se rapporter à n'importe quelle caractéristique mesurable ou observable résultant de l'interaction entre les gènes et l'environnement, y compris la couleur des yeux, la taille, le poids, certaines maladies ou conditions médicales, voire même la réponse à un traitement spécifique.
Il est important de noter que deux individus ayant le même génotype (c'est-à-dire la même séquence d'ADN) ne seront pas nécessairement identiques dans leur phénotype, car des facteurs environnementaux peuvent influencer l'expression des gènes. De même, des individus avec des génotypes différents peuvent partager certains traits phénotypiques en raison de similitudes dans leurs environnements ou dans d'autres facteurs non génétiques.
Un allèle est une forme alternative d'un gène donné qui est localisé à la même position (locus) sur un chromosome homologue. Les allèles peuvent produire des protéines ou des ARNm avec des séquences différentes, entraînant ainsi des différences phénotypiques entre les individus.
Les gènes sont des segments d'ADN qui contiennent les instructions pour la production de protéines spécifiques ou pour la régulation de l'expression génique. Chaque personne hérite de deux copies de chaque gène, une copie provenant de chaque parent. Ces deux copies peuvent être identiques (homozygotes) ou différentes (hétérozygotes).
Les allèles différents peuvent entraîner des variations subtiles dans la fonction protéique, ce qui peut se traduire par des différences phénotypiques entre les individus. Certaines de ces variations peuvent être bénéfiques, neutres ou préjudiciables à la santé et à la survie d'un organisme.
Les allèles sont importants en génétique car ils permettent de comprendre comment des caractères héréditaires sont transmis d'une génération à l'autre, ainsi que les mécanismes sous-jacents aux maladies génétiques et aux traits complexes.
La progression d'une maladie, également appelée évolution de la maladie, se réfère à la manifestation temporelle des stades ou étapes d'une maladie chez un patient. Il s'agit essentiellement de la détérioration continue ou de l'aggravation d'un trouble médical au fil du temps, qui peut entraîner une augmentation de la gravité des symptômes, une déficience accrue, une invalidité et, éventuellement, la mort. La progression de la maladie est généralement mesurée en termes de déclin fonctionnel ou de dommages aux organes affectés. Elle peut être influencée par divers facteurs, notamment l'âge du patient, la durée de la maladie, le traitement et les comorbidités sous-jacentes. Le suivi de la progression de la maladie est crucial pour évaluer l'efficacité des interventions thérapeutiques et pour la planification des soins futurs.
Le Conseil Génétique est une branche de la médecine qui consiste en des consultations avec un professionnel de santé qualifié, souvent un conseiller en génétique, pour aider les individus, les familles et les couples à comprendre les risques de transmission ou de développement d'une maladie liée à l'hérédité. Il fournit des informations sur la probabilité qu'une personne hérite ou transmette une maladie génétique, en se basant sur l'analyse de leur arbre généalogique, leurs antécédents médicaux et, si nécessaire, des tests génétiques.
Le conseil génétique vise également à assister les gens dans la prise de décisions éclairées concernant leur santé reproductive, y compris le diagnostic prénatal et les options de gestion de la fertilité. Il offre un soutien psychologique et un suivi continus pour faire face aux défis émotionnels liés à la compréhension et à l'acceptation des résultats.
En plus d'être bénéfique sur le plan individuel, le conseil génétique joue également un rôle crucial dans la recherche médicale et la santé publique en contribuant aux études épidémiologiques, à l'identification des facteurs de risque et à l'élaboration de stratégies de prévention pour les maladies héréditaires.
La liaison génétique est un phénomène dans lequel des gènes ou des marqueurs génétiques spécifiques situés à proximité les uns des autres sur un chromosome ont tendance à être hérités ensemble au cours de la méiose, car il est moins probable qu'ils soient séparés par recombinaison génétique. Plus la distance entre deux gènes ou marqueurs est petite, plus ils sont susceptibles d'être co-transmis et donc considérés comme étant liés. La mesure de cette liaison est exprimée en unités de carte génétique, telles que les centimorgans (cM), qui représentent environ un échange réciproque par recombinaison sur 100 meioses.
Ce concept est fondamental dans la cartographie génétique et l'analyse de l'expression des gènes, ainsi que dans l'identification des mutations causales de maladies monogéniques et complexes. Il permet également d'identifier des susceptibilités génétiques à certaines conditions médicales ou traits, ce qui peut conduire à un counseling génétique plus précis et à une médecine personnalisée.
Les composés nitrés sont une classe de composés organiques ou inorganiques qui contiennent un groupe fonctionnel avec une formule générale -NO2, -N=O ou -ONO. Dans le contexte médical, les composés nitrés les plus couramment mentionnés sont des composés organiques qui contiennent un ou plusieurs groupes fonctionnels -NO2. Les composés nitrés les plus connus en médecine sont les nitrates et les nitrites, qui sont largement utilisés dans le traitement de diverses affections cardiovasculaires.
Les nitrates sont des sels ou des esters d'acides nitriques avec une formule générale R-ONO2. Les exemples courants de nitrates comprennent le nitrate de sodium (NaNO3) et le nitrate de potassium (KNO3). Lorsqu'ils sont métabolisés dans le corps, les nitrates se décomposent en monoxyde d'azote (NO), un puissant vasodilatateur qui améliore la circulation sanguine et réduit la pression artérielle.
Les nitrites sont des sels ou des esters d'acides nitreux avec une formule générale R-ONO. Les exemples courants de nitrites comprennent le nitrite de sodium (NaNO2) et le nitrite d'amyle (CH3)2CH-ONO). Dans le passé, les nitrites étaient utilisés comme vasodilatateurs dans le traitement de l'angine de poitrine. Cependant, en raison du développement de médicaments plus sûrs et plus efficaces, leur utilisation est maintenant limitée.
En général, les composés nitrés sont utilisés dans le traitement des maladies cardiovasculaires pour améliorer la circulation sanguine, réduire la pression artérielle et prévenir l'angine de poitrine. Cependant, leur utilisation peut entraîner une tolérance et une dépendance, ce qui nécessite une surveillance étroite et des ajustements posologiques fréquents.
Le diagnostic précoce, dans le contexte médical, se réfère au processus d'identification et de confirmation d'une maladie ou d'un trouble chez un individu le plus tôt possible après l'apparition des symptômes. Il s'agit d'une approche proactive qui permet une intervention médicale rapide, améliorant ainsi les perspectives de traitement et de rétablissement du patient.
Le diagnostic précoce est particulièrement crucial dans le cas de maladies potentiellement mortelles ou dégénératives, telles que le cancer, la maladie d'Alzheimer, le Parkinson, etc., où un traitement rapide peut ralentir la progression de la maladie, réduire la gravité des symptômes et améliorer la qualité de vie du patient.
Cela peut être accompli grâce à une combinaison de facteurs, y compris des examens physiques réguliers, des antécédents médicaux détaillés, des tests de dépistage appropriés et une sensibilisation accrue aux signes et symptômes précoces d'une maladie. Le diagnostic précoce est un élément clé de la médecine préventive et peut contribuer de manière significative à des résultats cliniques positifs.
La détection hétérozygote en génétique médicale fait référence au processus de détermination de la présence d'une mutation génétique dans un seul des deux allèles d'un gène dans un individu. Dans le contexte de la médecine, cela est souvent important pour les maladies héréditaires à transmission autosomique récessive, où avoir une copie mutée du gène (être hétérozygote) ne provoque pas la maladie mais signifie que l'individu est un porteur de cette mutation. Si deux personnes hétérozygotes pour la même mutation ont un enfant ensemble, il y a une chance sur quatre qu'un enfant hérite d'une copie de chaque parent et développe donc la maladie. La détection des hétérozygotes est donc importante dans le conseil génétique et les tests prénataux pour ces conditions.
Une remnographie est un type d'examen d'imagerie médicale qui utilise une faible dose de radiation pour produire des images détaillées des structures internes du corps. Contrairement à une radiographie standard, une remnographie implique l'utilisation d'un milieu de contraste, comme un produit de contraste à base d'iode, qui est ingéré ou injecté dans le patient avant l'examen.
Le milieu de contraste permet aux structures internes du corps, telles que les vaisseaux sanguins, les organes creux ou les tissus mous, d'être plus visibles sur les images radiographiques. Cela peut aider les médecins à diagnostiquer une variété de conditions médicales, y compris les maladies gastro-intestinales, les maladies rénales et les troubles vasculaires.
Les remnographies sont généralement considérées comme sûres, bien que comme avec toute procédure médicale qui utilise des radiations, il existe un risque minimal de dommages aux tissus ou au matériel génétique. Les avantages potentiels d'un diagnostic précis et opportun sont généralement considérés comme dépassant ce faible risque.
Il est important de noter que les remnographies ne doivent être effectuées que lorsqu'elles sont médicalement nécessaires, car l'exposition répétée aux radiations peut augmenter le risque de dommages à long terme. Les médecins et les technologues en imagerie médicale prennent des précautions pour minimiser l'exposition aux radiations pendant les procédures de remnographie.
Le génotype, dans le contexte de la génétique et de la médecine, se réfère à l'ensemble complet des gènes héréditaires d'un individu, y compris toutes les variations alléliques (formes alternatives d'un gène) qu'il a héritées de ses parents. Il s'agit essentiellement de la constitution génétique innée d'un organisme, qui détermine en grande partie ses caractéristiques et prédispositions biologiques.
Les différences génotypiques peuvent expliquer pourquoi certaines personnes sont plus susceptibles à certaines maladies ou répondent différemment aux traitements médicaux. Par exemple, dans le cas de la mucoviscidose, une maladie génétique potentiellement mortelle, les patients ont généralement un génotype particulier : deux copies du gène CFTR muté.
Il est important de noter que le génotype ne définit pas entièrement les caractéristiques d'un individu ; l'expression des gènes peut être influencée par divers facteurs environnementaux et épigénétiques, ce qui donne lieu à une grande variabilité phénotypique (manifestations observables des traits) même entre les personnes partageant le même génotype.
Les troubles cognitifs sont un terme général utilisé pour décrire les changements dans le fonctionnement mental et cognitif qui affectent votre capacité à traiter et à comprendre l'information. Ils peuvent affecter la mémoire, le raisonnement, l'attention, la perception, le langage, la planification et le jugement. Les troubles cognitifs peuvent être causés par divers facteurs, tels que des maladies dégénératives du cerveau (comme la maladie d'Alzheimer), des lésions cérébrales, des infections cérébrales ou des désordres mentaux. Les symptômes peuvent varier en fonction de la cause sous-jacente et peuvent inclure une perte de mémoire, une confusion, une difficulté à résoudre des problèmes, une diminution de l'attention et de la concentration, une désorientation dans le temps et l'espace, et des changements de personnalité ou de comportement. Les troubles cognitifs peuvent avoir un impact significatif sur la capacité d'une personne à fonctionner dans sa vie quotidienne et peuvent nécessiter des soins et une prise en charge spécialisés.
La dégénérescence nerveuse est un terme général utilisé en médecine pour décrire une condition où les nerfs du corps se détériorent ou se décomposent. Cela peut se produire en raison de divers facteurs, tels que des maladies, des traumatismes, l'âge ou des habitudes malsaines.
La dégénérescence nerveuse peut affecter n'importe quel type de nerfs dans le corps, y compris les nerfs sensoriels (qui transmettent des sensations telles que la douleur, le toucher et la température), les nerfs moteurs (qui contrôlent les mouvements musculaires) et les nerfs autonomes (qui régulent les fonctions automatiques du corps telles que la fréquence cardiaque, la pression artérielle et la digestion).
Les symptômes de la dégénérescence nerveuse varient en fonction de la zone affectée et peuvent inclure des douleurs, des picotements, une faiblesse musculaire, une perte d'équilibre, une vision floue, des problèmes auditifs, des difficultés à avaler ou à parler, et une perte de contrôle de la vessie ou des intestins.
Le traitement de la dégénérescence nerveuse dépend de la cause sous-jacente. Dans certains cas, il peut être possible de ralentir ou d'arrêter la progression de la maladie grâce à des médicaments, une thérapie physique, une intervention chirurgicale ou d'autres traitements. Cependant, dans d'autres cas, la dégénérescence nerveuse peut être irréversible et entraîner des dommages permanents aux nerfs.
La cartographie chromosomique est une discipline de la génétique qui consiste à déterminer l'emplacement et l'ordre relatif des gènes et des marqueurs moléculaires sur les chromosomes. Cette technique utilise généralement des méthodes de laboratoire pour analyser l'ADN, comme la polymerase chain reaction (PCR) et la Southern blotting, ainsi que des outils d'informatique pour visualiser et interpréter les données.
La cartographie chromosomique est un outil important dans la recherche génétique, car elle permet aux scientifiques de comprendre comment les gènes sont organisés sur les chromosomes et comment ils interagissent entre eux. Cela peut aider à identifier les gènes responsables de certaines maladies héréditaires et à développer des traitements pour ces conditions.
Il existe deux types de cartographie chromosomique : la cartographie physique et la cartographie génétique. La cartographie physique consiste à déterminer l'emplacement exact d'un gène ou d'un marqueur sur un chromosome en termes de distance physique, exprimée en nucléotides. La cartographie génétique, quant à elle, consiste à déterminer l'ordre relatif des gènes et des marqueurs sur un chromosome en fonction de la fréquence de recombinaison entre eux lors de la méiose.
En résumé, la cartographie chromosomique est une technique utilisée pour déterminer l'emplacement et l'ordre relatif des gènes et des marqueurs moléculaires sur les chromosomes, ce qui permet aux scientifiques de mieux comprendre comment les gènes sont organisés et interagissent entre eux.
L'autophagie est un processus cellulaire naturel qui dégrade et recycle les composants intracellulaires, tels que les protéines endommagées et les organites, dans la cellule. Ce processus permet à la cellule de maintenir l'homéostasie et de s'adapter aux changements environnementaux et métaboliques. Dans des conditions normales, l'autophagie se produit à un rythme de base, mais elle peut être induite en réponse au stress cellulaire, telle que la privation de nutriments ou l'hypoxie.
Le processus d'autophagie implique plusieurs étapes:
1. L'isolement des composants cytoplasmiques à dégrader dans une vésicule membranaire appelée autophagosome.
2. La fusion de l'autophagosome avec une lysosome, une structure cellulaire contenant des enzymes digestives qui décomposent les composants organiques.
3. La dégradation des composants cytoplasmiques par les enzymes lysosomales dans un environnement acide.
4. Le recyclage des produits de dégradation pour la synthèse de nouveaux macromolécules et pour fournir de l'énergie à la cellule.
L'autophagie joue un rôle important dans le maintien de la santé cellulaire et a été impliquée dans plusieurs processus physiologiques, tels que le développement embryonnaire, la différenciation cellulaire et la réponse immunitaire. Des anomalies dans l'autophagie ont également été associées à plusieurs maladies, telles que les maladies neurodégénératives, les maladies cardiovasculaires, le cancer et les infections virales.
En résumé, l'autophagie est un processus cellulaire important qui permet la dégradation et le recyclage des composants cytoplasmiques dans la cellule. Il joue un rôle crucial dans le maintien de la santé cellulaire et a été impliqué dans plusieurs maladies lorsque les anomalies se produisent.
 Maladie de Huntington
Maladie de Huntington
Robert Huntington (orientaliste)
Huntington
Loi allemande sur la stérilisation forcée du 14 juillet 1933
Ganglions de la base
Saison 3 de Revenge
G10
Anticipation (génétique)
Cara Lott
Patricia Tuckwell
Vincent Astor
Expressivité (génétique)
Jean-Marc Daumas
Kinésine
Évolution du cerveau
Association française contre les myopathies
Procès des sorcières de Salem
Elena Cattaneo
Woody Guthrie
Huntingtine
Dominance (génétique)
Mutations (roman)
Thérapie cellulaire
Michael Jones (chanteur)
Laetitia Carton
Maladie génétique
Eve Babitz
André Parent
Nina Webster
Nathalie Cartier-Lacave
Maladie de Huntington - Wikipedia
 Fabrique de savoirs - L'imagerie préclinique multimodale éclaire le développement de la maladie de Huntington
Fabrique de savoirs - L'imagerie préclinique multimodale éclaire le développement de la maladie de Huntington
 Qu'est-ce que la maladie de Huntington ?
Qu'est-ce que la maladie de Huntington ?
 La maladie de Huntington serait visible dès le stade embry... - Top Santé
La maladie de Huntington serait visible dès le stade embry... - Top Santé
 Evaluation préclinique d'une thérapie cellulaire de la maladie de Huntington fondée sur l'utilisation de cellules souches...
Evaluation préclinique d'une thérapie cellulaire de la maladie de Huntington fondée sur l'utilisation de cellules souches...
 Appels à projets Maladie de Huntington - Fondation Maladies Rares
Appels à projets Maladie de Huntington - Fondation Maladies Rares
 Souffrant de la maladie de Huntington, mais plus jamais 'cachés'! - ZENIT - Francais
Souffrant de la maladie de Huntington, mais plus jamais 'cachés'! - ZENIT - Francais
 Taube offre 3 M $ pour la recherche sur la maladie de Huntington aux Etats-Unis - The Times of Israël
Taube offre 3 M $ pour la recherche sur la maladie de Huntington aux Etats-Unis - The Times of Israël
 maladie de huntington | Agence Lusso
maladie de huntington | Agence Lusso
 Programme colloque maladie de Huntington - RESPECTS 73
Programme colloque maladie de Huntington - RESPECTS 73
 Maladie de Huntington : un espoir avec l'oligonucléotide antisens !
Maladie de Huntington : un espoir avec l'oligonucléotide antisens !
 Dingdingdong - ICS Maladie de Huntington - La puissance de l'idée
Dingdingdong - ICS Maladie de Huntington - La puissance de l'idée
 Pharmacie Parent SPRL : Home > Solutions A-Z - Maladie de...
Pharmacie Parent SPRL : Home > Solutions A-Z - Maladie de...
 Centre de compétence de la maladie de Huntington - Martinique - ESMARA
Centre de compétence de la maladie de Huntington - Martinique - ESMARA
 Vers un traitement de la maladie de Huntington - Fondation Brain Canada
Vers un traitement de la maladie de Huntington - Fondation Brain Canada
Les Différentes Étapes de la Maladie de Huntington : Comprendre la Progression
 HDBuzz - Actualités à propos de la recherche sur la maladie de Huntington.
HDBuzz - Actualités à propos de la recherche sur la maladie de Huntington.
HDBuzz - Actualités à propos de la recherche sur la maladie de Huntington.
Résultats de recherche | Page 8 | CNSA
 À mesure que la maladie progresse (démence, SLA, SP, maladie de Parkinson ou maladie de Huntington) : Chapitre 2 : Vivre avec...
À mesure que la maladie progresse (démence, SLA, SP, maladie de Parkinson ou maladie de Huntington) : Chapitre 2 : Vivre avec...
À mesure que la maladie progresse (démence, SLA, SP, maladie de Parkinson ou maladie de Huntington) : Résumé du module:...
Institut de biologie François Jacob - Retour sur la première journée scientifique de PASREL-imagerie
 Explore Stem Cells | Eurostemcell
Explore Stem Cells | Eurostemcell
 COVID-19 : Du stress jusqu'à la pointe des cheveux | santé log
COVID-19 : Du stress jusqu'à la pointe des cheveux | santé log
 Maladie de Huntington - Troubles neurologiques - Édition professionnelle du Manuel MSD
Maladie de Huntington - Troubles neurologiques - Édition professionnelle du Manuel MSD
 hugocallens
hugocallens
 Recherche | CHUM
Recherche | CHUM
Maladie orpheline
 Réunion d'information - Huntington
Réunion d'information - Huntington
 theses.fr - Ecole normale supérieure (Paris ; 1985-....). Laboratoire de sciences cognitives et psycholinguistique
theses.fr - Ecole normale supérieure (Paris ; 1985-....). Laboratoire de sciences cognitives et psycholinguistique
Parkinson7
- Certains cas de démence résultent d'une autre maladie neurodégénérative comme celle de Huntington ou celle de Parkinson. (mondeuil.ca)
- La maladie de Parkinson. (mondeuil.ca)
- Le rythme de l'évolution de la maladie de Parkinson varie d'une personne à l'autre. (mondeuil.ca)
- Dans certaines familles, la maladie de Parkinson est héréditaire. (mondeuil.ca)
- Pour un complément d'information sur la maladie de Parkinson, consultez le site de Parkinson Canada. (mondeuil.ca)
- Ce projet existe déjà pour des maladies comme le cancer, la SLA, la maladie de Parkinson, Huntington, la grippe et bien d'autres. (zataz.com)
- Le médicament SAGE-718, est également à l'étude pour les troubles cognitifs légers chez les patients atteints de la maladie de Huntington, l'indication principale du médicament, et la maladie de Parkinson. (medscape.com)
D'Alzheimer10
- Le déclin cognitif, possiblement associé à des pathologies du système nerveux central comme la maladie d'Alzheimer, peut conduire les personnes à avoir souvent le sentiment de perdre des objets alors qu'ils les ont simplement égarés. (cnsa.fr)
- La maladie d'Alzheimer. (mondeuil.ca)
- Pour un complément d'information sur la maladie d'Alzheimer, consultez le site de la Société Alzheimer du Canada. (mondeuil.ca)
- Je suis plantée devant l'ordinateur, à un clic de découvrir si je suis porteuse d'«anomalies» génétiques susceptibles d'augmenter le risque de développer la maladie d'Alzheimer - ma hantise. (selection.ca)
- Sans parler des conséquences sociales ou psychologiques que peut entraîner la révélation de certaines informations, comme annoncer à un père qu'il n'est pas le géniteur de son enfant, ou apprendre que l'on est prédisposé au cancer du sein ou à la maladie d'Alzheimer. (selection.ca)
- Etats-Unis - Les résultats précoces d'un petit essai évaluant un médicament expérimental, le SAGE-718, montrent qu'il peut améliorer les fonctions exécutives chez les patients atteints de troubles cognitifs légers et de démence légère associée à la maladie d'Alzheimer (MA) et qu'il est bien toléré. (medscape.com)
- Dans l'essai ouvert de phase 2a LUMINARY , les chercheurs ont recruté 26 patients âgés de 50 à 80 ans atteints de la maladie d'Alzheimer et présentant un déficit cognitif léger. (medscape.com)
- Nous savons que dans la maladie d'Alzheimer et d'autres maladies neurodégénératives, il y a un changement dans la cognition, la capacité de penser, la capacité de faire des choses », a souligné le Dr Koenig. (medscape.com)
- La signification clinique réelle des résultats de recherche générés dans des environnements hautement contrôlés est une question importante qui fait actuellement l'objet de discussions dans le domaine de la maladie d'Alzheimer », a-t-il ajouté. (medscape.com)
- Sage Therapeutics prévoit de lancer un essai contrôlé contre placebo de phase 2b plus tard cette année afin d'étudier SAGE-718 chez davantage de patients atteints de la maladie d'Alzheimer et sur une plus longue période. (medscape.com)
Rares16
- Il existe d'ailleurs des formes plus rares de la maladie précoces (moins de 21 ans), ou au contraire tardives (de 50 à 80 ans). (wikipedia.org)
- La Fondation Maladies Rares bénéficie du soutien déterminant de l'AFM Téléthon. (fondation-maladiesrares.org)
- La Fondation a pour but de favoriser la conduite de projets de recherche et d'excellence scientifique ainsi que le partage et la diffusion des connaissances dans le domaine des maladies rares. (fondation-maladiesrares.org)
- Je voudrais étendre mes salutations à toutes les personnes qui, dans leur corps et dans leur vie, portent les signes de cette maladie, ainsi qu'à ceux qui souffrent d'autres pathologies dites rares. (zenit.org)
- Albert tente depuis des années de sensibiliser la société aux maladies rares, de favoriser un diagnostic rapide, et d'obtenir des soins adaptés. (radiorg.be)
- Pour des maladies rares de type Huntington, la connaissance des spécificités de la maladie et des techniques de suivi sont des éléments critiques. (radiorg.be)
- Cordes Tolosannes (82) l'association des Maladies Orphelines Midi-pyrenees a pour but d'effectuer des demarches en Midi-Pyrenees, de financer des actions, de mettre en place une communication appropriee et de collecter les cartouches d'encre en faveur de la recherche sur les maladies rares. (boussole-fr.com)
- Aide et soutien aux personnes atteintes de maladies genetiques rares du systeme immunitaire, deficits immunitaires primitifs (DIP). (boussole-fr.com)
- En partenariat avec l'équipe du centre de compétence de la maladie de Huntington - CHU de Clermont-Ferrand, la Résidence Mutualiste Transverse et l'Equipe Relais Handicaps Rares du Puy-de-Dôme, la délégation régionale AHF Auvergne-Rhône-Alpes vous convie à la journée de rencontre des malades Huntington et de leur famille. (huntington.fr)
- Alors que seulement 5 % des maladies rares disposent d'un traitement médicamenteux efficace, on déplore pourtant l'indisponibilité de beaucoup d'entre eux au Maroc. (yabiladi.com)
- De plus, de nombreuses maladies rares n'ont pas le statut d'affection de longue durée (ALD), garantissant prise en charge et remboursement systématiques des soins. (yabiladi.com)
- Cet émiettement ne favorise pas la viabilité financière à long terme de la couverture médicale dans son ensemble car il ne permet pas de répartir la charge financière qu'induit la cherté des soins dans les maladies rares sur toute la population. (yabiladi.com)
- Pour toutes ces raisons, les patients atteints de maladies rares font face à des coûts prohibitifs qui les font renoncer pour la plupart à la plénitude des soins et thérapeutiques nécessaires. (yabiladi.com)
- Les maladies dites rares touchent chacune par définition un nombre restreint de personnes, moins d'une personne sur 2 000 dans la population. (yabiladi.com)
- Ce forum a pour objectifs de lancer un appel urgent en faveur d'une amélioration de la prise en charge des maladies rares dans notre pays en leur octroyant le statut d'affection de longue durée et en assurant une pleine disponibilité et un remboursement des médicaments orphelins. (yabiladi.com)
- Extrêmement diverses, 3 maladies rares sur 4 se déclenchent dans l'enfance mais certaines attendent 30, 40 ou 50 ans avant de se déclarer. (yabiladi.com)
Traitements7
- Cette phase est relativement mal connue et il serait intéressant de disposer de biomarqueurs précoces pour mieux comprendre la progression de la maladie et développer de futurs traitements. (cea.fr)
- Ce don appuiera un nouveau programme mettant en place trois groupes de recherche médicale et, pour la première fois, introduira des thérapies de cellules souche et d'édition de gènes pour déterminer des traitements et pour apporter des remèdes potentiels permettant de soigner la maladie de Huntington, a fait savoir le groupe de philanthropes dans un communiqué. (timesofisrael.com)
- Le don de trois millions de dollars vient soutenir l'objectif de Taube Philanthropies de trouver des traitements et des remèdes pour venir à bout des maladies neuro-dégénératives. (timesofisrael.com)
- Aujourd'hui, les traitements permettent de soulager les symptômes, mais pas de ralentir la progression de la maladie ou de stopper sa survenue. (sante-sur-le-net.com)
- Ensemble, ces travaux aideront à tirer au clair les mécanismes d'action des GM1 dans cette maladie, tout en accélérant le développement de traitements à base de gangliosides. (braincanada.ca)
- Certains traitements peuvent ralentir la progression de la maladie ou aider à en contrôler les symptômes, mais il n'existe actuellement aucun remède. (mondeuil.ca)
- On se sert de ces modèles pour comprendre comment fonctionnent ces maladies et ainsi trouver des traitements. (univ-evry.fr)
D'autres2
- Elles ont pour cela étudié des embryons humains, pour certains avec l'hérédité de la maladie et pour d'autres, sans . (topsante.com)
- D'autres régions ont signalé que certains individus et dirigeants religieux pourraient juger acceptable le clonage à des fins de reproduction dans certains cas comme la stérilité non traitable, ou pour éviter une maladie génétique héréditaire. (who.int)
Malades5
- La maladie de Huntington est une maladie rare dont la prévalence, stable, se maintient entre 5 et 7 malades pour 100 000 au sein de la population caucasienne. (wikipedia.org)
- Stanford effectue déjà une recherche sur les cellules souches et espère commencer les essais cliniques auprès des malades de Huntington rapidement, explique le communiqué. (timesofisrael.com)
- Pour mener à bien cet essai clinique de phase 1-2a , les chercheurs supervisés par la professeure Sarah Tabrizi et parrainés par Ionis Pharmaceuticals, ont recruté 46 malades diagnostiqués à un stade précoce de la maladie et âgés de 25 à 65 ans. (sante-sur-le-net.com)
- Et on doit beaucoup veiller à la nutrition (les malades Huntington ont besoin de +/- 3.000 calories par jour alors qu'ils ont de grandes difficultés de déglutition). (radiorg.be)
- En partenariat avec l'équipe du centre de référence d'Angers des maladies neuro-génétiques, la délégation régionale AHF Bretagne - Pays de la Loire organise le samedi 16 octobre 2021 une journée de rencontre des malades Huntington et leurs familles au Centre Ethic Etapes à Angers. (huntington.fr)
Cerveau10
- En 2018, une étude met en évidence, sur des modèles murins de la maladie de Huntington, une vulnérabilité accrue du corps calleux - une structure de la substance blanche connectant les deux hémisphères du cerveau. (cea.fr)
- La maladie de Huntington est une pathologie neuro-dégénérative génétique qui cause la rupture progressive des cellules nerveuses du cerveau. (timesofisrael.com)
- La maladie de Huntington est une maladie héréditaire dans laquelle les cellules nerveuses de certaines régions du cerveau meurent. (pharmacieparent.be)
- La maladie de Huntington est un trouble héréditaire qui entraîne l'anéantissement progressif des parties du cerveau responsables du contrôle des mouvements, du raisonnement et des émotions. (braincanada.ca)
- Les maladies et troubles neurocognitifs affectent le cerveau de la personne et tout son être. (mondeuil.ca)
- Cette maladie évolutive attaque le cerveau et le système nerveux. (mondeuil.ca)
- aussi appelée maladie de Lou Gehrig ou maladie des motoneurones, affecte la capacité du cerveau à communiquer avec les muscles du corps, ce qui entraîne une paralysie progressive. (mondeuil.ca)
- est une maladie chronique du système immunitaire qui affecte le système nerveux central, y compris le cerveau, la moelle épinière et le nerf optique. (mondeuil.ca)
- Tour d'horizon sur la Recherche et les avancées concernant la Maladie de Huntington par Emmanuel Brouillet, directeur de Recherches au CNRS et discussion avec Laurie Galvan, maître de conférence à l'Université de Poitiers, à l'occasion de la semaine du Cerveau le 18 mars 2021 à Poitiers (Espace Mendes France). (huntington.fr)
- Puis fin 2008 il se spécialise en neuroscience et intègre l'Institut Curie où il travaille plus particulièrement sur la maladie de Huntington (maladie génétique du cerveau). (univ-evry.fr)
Mutation7
- La maladie de Huntington est causée par une mutation génétique du gène codant la protéine huntingtine . (sante-sur-le-net.com)
- La maladie de Von Hippel-Lindau (VHL) est une maladie génétique rare due à une mutation du gène VHL. (cnsa.fr)
- La maladie de Huntington résulte d'une mutation du gène de l' huntingtin e ( HTT ) (sur le chromosome 4), avec une expansion anormale de triplets CAG de l'ADN qui code pour l'acide aminé glutamine. (msdmanuals.com)
- Porteur asymptomatique de la maladie de Huntington (MH), Albert Counet a transmis un accroissement de la mutation du gène à son fils Cédric, décédé en 2012 à 42 ans. (radiorg.be)
- L'annonce que l'on est porteur de la mutation génétique responsable de la maladie de Huntington, dans le cadre de la consultation génétique, correspond à un profond bouleversement du point de vue personnel. (espace-ethique.org)
- C'est-à-dire ceux qui recherchent chez une personne donnée des maladies liées à l'altération, ou la mutation, d'un seul gène, comme la maladie de Huntington. (selection.ca)
- On essaye de générer des cellules qui ont une mutation génétique qui cause une maladie chez l'homme et entraîne une perte rapide de la vision. (univ-evry.fr)
Traitement11
- Les premiers signes cliniques de la maladie de Huntington n'apparaissent qu'à l'âge adulte et aucun traitement ne permet aujourd'hui de guérir cette affection neurodégénérative, rare et héréditaire, ni même de ralentir son évolution. (cea.fr)
- HD-SCT (HD-Stem Cell Therapy) traite de l'analyse approfondie du potentiel thérapeutique de hPSC comme une source de greffon pour le traitement de la maladie de Huntington. (anr.fr)
- Il n'y a ni traitement ni médicament approuvé pour réduire la progression de la maladie. (timesofisrael.com)
- Nous sommes ravis d'aller de l'avant pour un essai mondial de phase 3 sponsorisé par F. Hoffmann-La Roche, qui a autorisé le médicament, afin de déterminer si un traitement antisens intrathécal régulier ralentit la progression clinique de la maladie. (sante-sur-le-net.com)
- Pour l'instant, il est possible d'atténuer les symptômes, mais il n'existe aucun traitement capable de retarder la maladie ou d'en arrêter la progression. (braincanada.ca)
- Il y a donc un besoin criant pour un traitement modificateur de la maladie. (braincanada.ca)
- À l'aide de modèles murins, on a démontré qu'il est possible d'atténuer radicalement les symptômes de la maladie de Huntington et d'en ralentir le processus neurodégénératif, une technique de traitement qui semble prometteuse. (braincanada.ca)
- Dans le cadre de ce projet, les chercheurs continueront d'approfondir leur connaissance des gangliosides afin de mettre au jour un traitement de la maladie de Huntington. (braincanada.ca)
- Pour Huntington, il n'existe aucun traitement. (radiorg.be)
- La meilleure connaissance de ces troubles permet une meilleure prise en compte et un traitement plus efficace, en particulier par l'interféron bêta indiqué dès les phases précoces de la maladie. (academie-medecine.fr)
- Nous croyons que chaque don que nous recevons est précieux et nous nous sommes engagés à mettre chaque cadeau à travailler pour trouver un traitement utile et de fournir un soutien et des services aux personnes touchées par la maladie de Huntington. (huntingtonsociety.ca)
Progression3
- La progression de la maladie suit un rythme et une forme extrêmement différents d'un individu à l'autre. (wikipedia.org)
- Or, l'atrophie de ces structures cérébrales est le principal biomarqueur utilisé pour évaluer la progression de la maladie chez l'homme. (cea.fr)
- Ils ont pour objectif d'identifier les mécanismes des gangliosides thérapeutiques, de les mesurer dans des échantillons humains et de vérifier s'ils se prêtent bien à une utilisation comme biomarqueur de la progression de la maladie de Huntington. (braincanada.ca)
Recherche sur les maladies1
- Des chercheurs de l'école de médecine de l'université de Stanford et du centre Taube-Koret de recherche sur les maladies neuro-dégénératives rattaché au Gladstone Institutes participeront à des essais clinique au Centre de la mémoire et de la vieillesse de l'université de Californie à San Francisco (UCSF). (timesofisrael.com)
Atteints3
- Enfin, ce projet bénéficie de l'aide inestimable de patients atteints de la maladie de Huntington et de leurs proches qui contribuent à l'atteinte des objectifs en fournissant des échantillons et des données. (braincanada.ca)
- Ces recherches fascinantes sont un premier pas vers des essais cliniques chez les patients atteints de la maladie de Huntington. (braincanada.ca)
- Ces symptômes prédisposent les patients aux idées suicidaires et au suicide, qui sont beaucoup plus fréquents chez les patients atteints de la maladie de Huntington que dans la population générale. (msdmanuals.com)
20221
- Le service de neurologie de l'hôpital de La Timone et l'antenne locale AHF de la région PACA organisent une réunion d'information sur la maladie de Huntington le samedi 24 septembre 2022, de 9 h à 12 h, à l'Amphithéâtre HA2 de l'hôpital La Timone, 264 Rue Saint-Pierre, à Marseille. (huntington.fr)
L'homme4
- Adapter ce protocole d'imagerie multimodal en clinique chez l'homme permettrait de définir des biomarqueurs plus robustes de la phase pré-symptomatique de la maladie pour une meilleure évaluation de thérapeutiques ciblées. (cea.fr)
- Pour Jésus, la maladie n'a jamais été un obstacle pour rencontrer l'homme, mais plutôt le contraire. (zenit.org)
- L'organisation Taube Philanthropies, fondée par l'homme d'affaires juif américain Tad Taube, a indiqué faire don de trois millions de dollars pour soutenir une collaboration entre l'école de médecine de l'Université de Stanford et le Gladstone Institutes en Californie, collaboration qui s'intéressera à la recherche associée à la maladie de Huntington. (timesofisrael.com)
- La maladie de Creutzfeld-Jacob est une maladie très rare, qui se manifeste chez l'homme suite à une contamination par un. (passeportsante.net)
Familles2
- Comprendre les différentes étapes de cette maladie est essentiel pour les patients, les familles et les professionnels de la santé afin de mieux gérer la condition et d'offrir un soutien approprié. (retraiteplus.be)
- La Ligue Huntington Francophone Belge est un groupe d'entraide et de soutien pour les familles confrontées à la maladie de Huntington dans la partie francophone de Belgique. (radiorg.be)
Parfois4
- Mise en garde médicale modifier - modifier le code - voir Wikidata (aide) La maladie de Huntington (parfois appelée chorée de Huntington) est une maladie héréditaire et rare, qui se traduit par une dégénérescence neurologique provoquant d'importants troubles moteurs, cognitifs et psychiatriques, et évoluant jusqu'à la perte d'autonomie puis la mort. (wikipedia.org)
- Le stade précoce de la maladie de Huntington est souvent caractérisé par des changements subtils et parfois négligeables. (retraiteplus.be)
- Quand on prend soin d'une personne atteinte d'une maladie cérébrale dégénérative, on vit des pertes et des deuils, parfois sur une longue période. (mondeuil.ca)
- Il vit cette maladie seul mais son entourage, notamment sa famille proche, participe à cette épreuve chronique aux conséquences socioprofessionnelles et familiales parfois dramatiques. (academie-medecine.fr)
Personnes5
- La maladie se développe chez des personnes âgées en moyenne de 40 à 50 ans. (wikipedia.org)
- Elle touche actuellement en France 18 000 personnes, ce qui fait d'elle une maladie dite rare. (topsante.com)
- C'est avec ces paroles que le pape François s'est adressé aux personnes affectées de la maladie de Huntington qui s'étaient réunis, accompagnées de leurs proches, ce jeudi 18 mai 2017 dans la Salle Paul VI, au Vatican. (zenit.org)
- Pendant trop longtemps, les peurs et les difficultés qui ont caractérisé la vie des personnes affectées par la maladie de Huntington ont créé autour d'elles des malentendus, des barrières et de véritables marginalisations. (zenit.org)
- Informations et forum de discussion pour les personnes concernees par la maladie de Huntington. (boussole-fr.com)
Troubles2
- La maladie commence par une phase cliniquement silencieuse (pré-symptomatique) à laquelle succèdent des troubles moteurs, psychologiques et cognitifs s'aggravant peu à peu. (cea.fr)
- Les symptômes de la maladie de Huntington étant souvent variables d'un malade à l'autre et très semblables à ceux d'une personne atteinte de troubles d'ordre uniquement psychologique, le diagnostic de la maladie de Huntington, est très difficile à poser. (retraiteplus.fr)
Huntingtine5
- Maladie de Huntington Inclusions nucléaires liées à la Huntingtine mutée (en jaune orangé) dans le cadre de la maladie de Huntington. (wikipedia.org)
- Les causes de la maladie de Huntington sont connues : la huntingtine devient toxique chez les porteurs de cette protéine mutante. (braincanada.ca)
- Le produit du gène, une protéine de grande taille appelée huntingtine, comporte une expansion de résidus polyglutamine qui s'accumulent dans les neurones et provoquent la maladie par des mécanismes inconnus. (msdmanuals.com)
- En effet, les équipes de Jean-Christophe Roux (équipe de Neurogénétique Humaine) du Centre de Génétique Médicale de Marseille* et Frédéric Saudou du Grenoble Institut Neurosciences** ont élaboré une stratégie originale en axant leurs travaux sur la huntingtine, protéine impliquée dans la maladie de Huntington. (asso.fr)
- L'activation de la protéine Huntingtine, qui lorsque mutée est responsable de la maladie de Huntington (MH), améliore la physiopathologie et les symptômes dans des souris modèles du syndrome de Rett. (asso.fr)
Qu'à1
- Tinne, qui est atteinte du syndrome d'Usher, est confrontée dans sa vie quotidienne aux problèmes de perte d'audition et de vision, ainsi qu'à des problèmes des démarches liés à sa maladie. (radiorg.be)
D'un1
- Il convient de l'appeler « maladie de Huntington » et non « chorée de Huntington », dans la mesure où le terme « chorée » désignait cette maladie seulement par l'un de ses symptômes moteurs (qui d'ailleurs peut manquer dans le tableau d'un malade), alors qu'elle provoque en réalité tout un faisceau de symptômes, lesquels constituent une maladie, pas une chorée. (wikipedia.org)
Soutien1
- Le stade avancé de la maladie de Huntington peut durer plusieurs années, et les patients deviennent de plus en plus dépendants des soins et du soutien de leurs proches ou de professionnels de la santé. (retraiteplus.be)
Stade8
- S'ensuit une dégradation progressive des fonctions cognitives aboutissant à la démence au stade final de la maladie. (retraiteplus.fr)
- Les premiers symptômes de la maladie de Huntington se manifestent par de légers changements physiques et éventuellement par des modifications sur le plan cognitif ou émotionnel pour évoluer petit à petit vers le stade avancé de la maladie. (retraiteplus.fr)
- La maladie de Huntington serait visible dès le stade embry. (topsante.com)
- La maladie de Huntington, qui entraîne une dégénérescence à l'âge adulte, généralement entre 30 et 50 ans, serait en fait présente dès le stade embryonnaire. (topsante.com)
- Les patients au stade précoce de la maladie sont généralement capables de maintenir une certaine autonomie et peuvent continuer à travailler et à participer à des activités quotidiennes. (retraiteplus.be)
- Au stade intermédiaire, les symptômes de la maladie de Huntington deviennent plus évidents et gênants. (retraiteplus.be)
- Le stade avancé de la maladie de Huntington est marqué par une détérioration significative de la fonction motrice et cognitive. (retraiteplus.be)
- Le stade terminal de la maladie de Huntington est la phase finale de la maladie, caractérisée par une dégradation grave de la fonction motrice et cognitive. (retraiteplus.be)
Paralysie1
- une sclérose cérébrale et une paralysie progressive de toutes les fonctions (leucodystrophie) … ou encore des mouvements incontrôlables et un affaiblissement intellectuel allant jusqu'à la démence (maladie de Huntington). (yabiladi.com)
Mouvements1
- Au début de la maladie, ces mouvements involontaires peuvent alors être interprétés comme de la nervosité, des tics, de la maladresse. (wikipedia.org)
Responsable1
- Un gène responsable de la maladie de Usher de type 1, repéré sur le chromosome 14 : des souris et des hommes. (inserm.fr)
Neurones1
- Dans la maladie de Huntington, on observe une atrophie du noyau caudé, les neurones inhibiteurs épineux moyens dans le striatum dégénèrent et les taux de neurotransmetteurs l'acide gamma-aminobutyrique (GABA) et de substance P diminuent. (msdmanuals.com)
Premiers1
- George Huntington, médecin généraliste du XIXe siècle originaire de Long Island (États-Unis), fut l'un des premiers à décrire les symptômes de la maladie qui portera par la suite son nom, lors d'une conférence prononcée le 15 février 1872. (wikipedia.org)
Incurable1
- C'est une maladie incurable. (braincanada.ca)
Plaques1
- La sclérose en plaques (SEP), maladie inflammatoire démyélinisante du système nerveux central, évolue par poussées. (academie-medecine.fr)
Dingdingdong1
- En créant Dingdingdong, nous sommes en train de franchir ce premier pas - tremblants de joie et d'inquiétude face à la tâche infinie, gorgée de vie, qui nous mènera non pas à la bonne définition de la maladie de Huntington, mais à la mise en œuvre de sa bonne composition. (dingdingdong.org)
D'information1
- Pour un complément d'information sur la maladie de Huntington, consultez le site de la Société Huntington du Canada. (mondeuil.ca)
Physiques1
- La maladie provoque des symptômes physiques, mentaux et psychologiques. (pharmacieparent.be)
Travaillons1
- Nous travaillons à développer des thérapies similaires pour les maladies neuro-dégénératives depuis de nombreuses années avec certains résultats prometteurs », dit le docteur Matthew Porteus, professeur associé de pédiatrie à Stanford. (timesofisrael.com)
Soutenir1
- L'Association Huntington France (AHF) propose 3 appels à projets pour soutenir la recherche française sur la maladie de Huntington. (fondation-maladiesrares.org)