Syndrome De Marfan
Protéines Microfilament
Ectopie Du Cristallin
Anévrysme Disséquant
Loeys-Dietz Syndrome
Microfibrilles
Anévrysme De L'Aorte Thoracique
Prolapsus De La Valve Mitrale
Chromosomes Humains De La Paire 15
Tissu
Généalogie
Thorax En Entonnoir
Elastin
Syndrome De Down
Syndrome Métabolique X
Mutation
Implantation Prothèse Vasculaire
Prolapsus De La Valve Tricuspide
Insuffisance Aortique
Le syndrome de Marfan est un trouble génétique des tissus conjonctifs qui affecte principalement le système cardiovasculaire, les yeux et le squelette. Cette maladie est causée par une mutation du gène FBN1 qui code pour la protéine fibrilline-1, essentielle à la formation des microfibrilles, composants structurels importants dans les tissus conjonctifs.
Les personnes atteintes de ce syndrome présentent typiquement une apparence allongée avec des membres longs et minces, un thorax en poire (thorax étroit à la base et élargi au niveau des épaules), une hypermobilité articulaire, une scoliose et/ou une cyphose, ainsi qu'un risque accru de déchirure ou de prolapsus de la valve mitrale dans le cœur. Les yeux peuvent également être affectés, entraînant des lentilles oculaires déplacées (ectopie lens) et un glaucome.
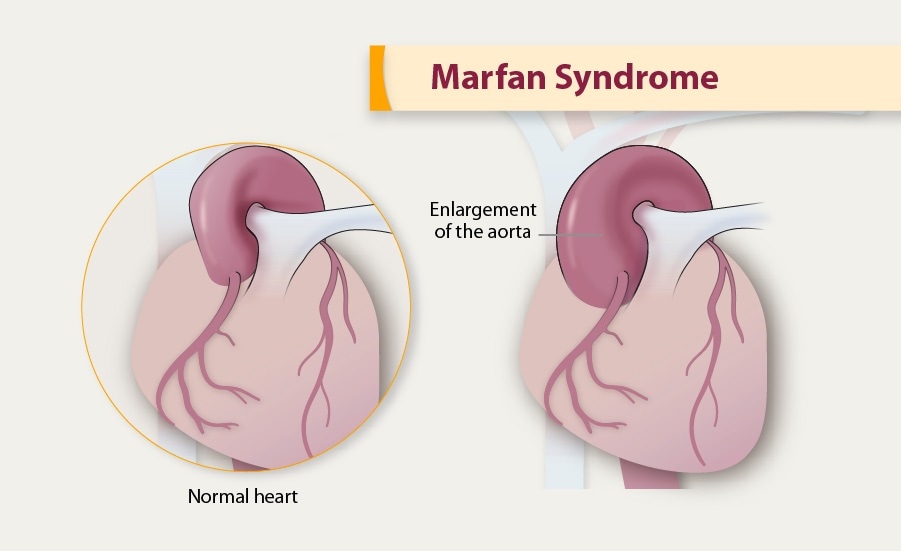
Le syndrome de Marfan peut également affecter les poumons, le système nerveux et la peau dans une certaine mesure. Les complications cardiovasculaires graves sont la principale cause de décès chez ces patients, notamment l'anévrisme et la dissection aortiques. Le diagnostic est généralement posé sur la base des critères cliniques de Ghent et peut être confirmé par un test génétique.
Le traitement du syndrome de Marfan implique une prise en charge multidisciplinaire, comprenant des soins cardiovasculaires, ophtalmologiques et orthopédiques spécialisés. Les médicaments bêta-bloquants sont souvent prescrits pour ralentir la progression de l'anévrisme aortique, tandis que la chirurgie peut être recommandée dans certains cas pour réparer ou remplacer les vaisseaux sanguins endommagés.
Un syndrome, dans le contexte médical, est un ensemble de symptômes ou de signes cliniques qui, considérés dans leur globalité, suggèrent l'existence d'une pathologie spécifique ou d'un état anormal dans le fonctionnement de l'organisme. Il s'agit essentiellement d'un ensemble de manifestations cliniques qui sont associées à une cause sous-jacente commune, qu'elle soit connue ou inconnue.
Un syndrome n'est pas une maladie en soi, mais plutôt un regroupement de signes et symptômes qui peuvent être liés à différentes affections médicales. Par exemple, le syndrome métabolique est un ensemble de facteurs de risque qui augmentent la probabilité de développer des maladies cardiovasculaires et du diabète de type 2. Ces facteurs comprennent l'obésité abdominale, l'hypertension artérielle, l'hyperglycémie à jeun et les taux élevés de triglycérides et de faibles taux de HDL-cholestérol.
La définition d'un syndrome peut évoluer avec le temps, alors que la compréhension des mécanismes sous-jacents s'améliore grâce aux recherches médicales et scientifiques. Certains syndromes peuvent être nommés d'après les professionnels de la santé qui ont contribué à leur identification ou à leur description, comme le syndrome de Down (trisomie 21) ou le syndrome de Klinefelter (XXY).
Il est important de noter que la présence d'un syndrome ne permet pas toujours d'établir un diagnostic définitif, car plusieurs affections médicales peuvent partager des symptômes similaires. Cependant, l'identification d'un syndrome peut aider les professionnels de la santé à orienter le diagnostic et le traitement vers des causes probables ou à fournir des informations sur le pronostic et la prise en charge globale du patient.
Les protéines microfilaments, également connues sous le nom de filaments d'actine, sont des structures fibreuses intracellulaires qui forment un réseau dynamique dans les cellules. Elles sont principalement composées de la protéine actine globulaire (G-actine) qui polymérise pour former des filaments rigides et flexibles (F-actine). Les microfilaments jouent un rôle crucial dans divers processus cellulaires, tels que la déformation cellulaire, le transport intracellulaire, la division cellulaire, la migration cellulaire et l'adhésion cellulaire. Ils interagissent avec d'autres protéines pour former des complexes protéiques qui régulent leur assemblage, leur désassemblage et leur organisation spatiale. Les médicaments qui ciblent les microfilaments peuvent affecter ces processus cellulaires et sont donc étudiés dans le cadre de diverses applications thérapeutiques.
Un anévrisme de l'aorte est une dilatation localisée et permanente de la paroi de l'aorte, qui est l'artère principale qui transporte le sang oxygéné loin du cœur vers les organes du corps. La dilatation se produit lorsque la paroi de l'aorte s'affaiblit et se dilate, ce qui peut entraîner une augmentation du diamètre de l'aorte à ce niveau.
Les anévrismes de l'aorte peuvent survenir dans n'importe quelle partie de l'aorte, mais ils sont les plus fréquents dans l'aorte abdominale, qui est la section de l'aorte située dans l'abdomen. Les anévrismes de l'aorte thoracique, qui se produisent dans la section de l'aorte située dans la poitrine, sont moins fréquents mais peuvent également être graves.
Les anévrismes de l'aorte peuvent entraîner des complications graves, telles que la rupture de l'anévrisme, qui peut être fatale. Les symptômes d'un anévrisme de l'aorte peuvent inclure une douleur abdominale ou thoracique sévère, une sensation de malaise, des nausées et des vomissements. Cependant, de nombreux anévrismes de l'aorte ne présentent aucun symptôme et sont découverts lors d'examens médicaux de routine.
Le traitement des anévrismes de l'aorte dépend de leur taille, de leur localisation et de la présence de symptômes. Les petits anévrismes asymptomatiques peuvent être surveillés par imagerie médicale régulière pour détecter toute augmentation de taille. Les anévrismes plus grands ou ceux qui présentent des symptômes peuvent nécessiter une intervention chirurgicale pour réparer ou remplacer la section affaiblie de l'aorte.
L'ectopie du cristallin, également connue sous le nom de luxation du cristallin, est une condition dans laquelle le lens naturel de l'œil se déplace de sa position normale. Dans des conditions normales, le cristallin est maintenu en place par les fibres suspensives qui s'attachent à la capsule postérieure du cristallin et à la paroi postérieure du sac vitréen.
Dans l'ectopie du cristallin, ces fibres suspensives sont affaiblies ou endommagées, ce qui entraîne le déplacement du cristallin vers l'avant ou vers l'arrière de l'œil. Cette condition peut être présente à la naissance (congénitale) ou peut se développer plus tard dans la vie en raison d'une blessure oculaire, d'une maladie oculaire sous-jacente ou d'un traumatisme.
Les symptômes de l'ectopie du cristallin peuvent inclure une vision floue, des halos autour des lumières vives, une sensibilité à la lumière et dans certains cas, une douleur oculaire ou une perte de vision soudaine. Le traitement dépend de la gravité de la condition et peut inclure des lunettes ou des lentilles de contact pour corriger la vision floue, ou une intervention chirurgicale pour retirer le cristallin endommagé et le remplacer par une lentille artificielle.
Un anévrisme disséquant est une affection vasculaire grave dans laquelle la paroi d'un vaisseau sanguin, généralement une artère, s'affaiblit et se dilate, créant ainsi une poche ou un sac. Cette poche peut alors se rompre, entraînant une hémorragie interne qui peut être fatale.
Les anévrismes disséquant se produisent le plus souvent dans l'aorte, la principale artère qui transporte le sang du cœur vers le reste du corps. Les facteurs de risque comprennent l'hypertension artérielle, le tabagisme, une maladie cardiovasculaire sous-jacente et certains troubles génétiques.
Les symptômes d'un anévrisme disséquant peuvent inclure une douleur thoracique ou abdominale sévère, des difficultés respiratoires, une sensation de faiblesse ou un engourdissement dans les bras ou les jambes, et une perte de conscience. Le traitement dépend de la gravité de l'anévrisme et peut inclure des médicaments pour abaisser la pression artérielle, une intervention chirurgicale pour réparer ou remplacer la section endommagée de l'artère, ou une endovasculaire moins invasive. procédure pour renforcer la paroi de l'artère.
Le syndrome de Loeys-Dietz est un trouble génétique qui affecte la façon dont le corps forme et maintient les tissus conjonctifs, qui sont les fibres qui soutiennent les organes, les vaisseaux sanguins et d'autres structures dans le corps. Il se caractérise par des anomalies vasculaires, y compris des anévrismes et des dissections aortiques, ainsi que des malformations craniofaciales, cutanées et squelettiques.
Les signes et symptômes du syndrome de Loeys-Dietz peuvent varier considérablement d'une personne à l'autre, mais ils comprennent souvent :
* Une hypertélorisme (yeux largement espacés)
* Un nez large et aplati
* Une fente palatine ou un palais creux
* Des oreilles malformées
* Une peau fine et élastique
* Des articulations hypermobiles
* Une scoliose ou une cyphose
* Des anévrismes et des dissections aortiques, qui peuvent entraîner des accidents vasculaires cérébraux, des douleurs thoraciques ou une insuffisance cardiaque
Le syndrome de Loeys-Dietz est causé par des mutations dans l'un des deux gènes TGFBR1 ou TGFBR2. Ces gènes fournissent des instructions pour la production d'une protéine qui joue un rôle important dans le développement et la réparation des tissus conjonctifs. Les mutations de ces gènes entraînent une activation excessive du signal TGF-β, ce qui perturbe la formation et le maintien des tissus conjonctifs.
Le syndrome de Loeys-Dietz est héréditaire et peut être transmis dans les familles selon un mode autosomique dominant, ce qui signifie qu'une seule copie du gène muté suffit pour que la maladie se manifeste. Les personnes atteintes de cette maladie ont un risque accru d'anévrismes et de dissections aortiques, il est donc important de surveiller régulièrement leur état cardiovasculaire. Le traitement du syndrome de Loeys-Dietz dépend des symptômes spécifiques et peut inclure des médicaments pour contrôler la pression artérielle, des interventions chirurgicales pour réparer les anévrismes ou les dissections aortiques, et une thérapie physique pour renforcer les muscles et améliorer la mobilité.
Les microfibrilles sont de minces structures filamenteuses qui mesurent généralement entre 10 à 12 nanomètres de diamètre et peuvent atteindre plusieurs micromètres de longueur. Elles sont principalement composées de glycosaminoglycanes (GAG) et de protéines, notamment de la famille des protéoglycanes et d'élastine. Les microfibrilles forment des réseaux tridimensionnels dans les tissus conjonctifs et élastiques, tels que la peau, les vaisseaux sanguins, les ligaments et les poumons. Elles jouent un rôle crucial dans l'organisation et la fonction mécanique des tissus en fournissant une structure de soutien, en régulant la distribution des cellules et en stockant des facteurs de croissance. Les microfibrilles sont également essentielles à la biogenèse de l'élastine, car elles servent de matrice sur laquelle les molécules d'élastine s'assemblent pour former des fibres élastiques fonctionnelles.
La dilatation pathologique est un terme médical qui se réfère à l'expansion ou au gonflement anormal d'une cavité ou d'un conduit dans le corps. Cela peut être dû à une variété de causes, y compris l'inflammation, l'infection, l'obstruction, la tumeur ou un traumatisme. La dilatation pathologique peut survenir dans divers organes et systèmes du corps, tels que les voies respiratoires, le tractus gastro-intestinal, les vaisseaux sanguins, le cœur, les reins et l'utérus.
Par exemple, la dilatation pathologique des bronches (dilatation des bronches) peut être observée dans certaines maladies pulmonaires telles que la bronchectasie, où les parois des bronches deviennent permanentes et anormalement larges. De même, la dilatation pathologique de l'estomac (dilatation gastrique) peut survenir en raison d'un blocage ou d'une obstruction de l'estomac, entraînant une expansion anormale de l'organe.
La dilatation pathologique peut causer des symptômes tels que la douleur, l'inconfort, des difficultés à avaler ou à respirer, des nausées et des vomissements, en fonction de l'emplacement et de la cause sous-jacente. Le traitement dépendra de la cause sous-jacente et peut inclure des médicaments, une intervention chirurgicale ou d'autres procédures thérapeutiques.
Un anévrisme de l'aorte thoracique est une dilatation localisée et focale de la paroi de l'aorte thoracique qui entraîne une augmentation de son diamètre de plus de 50%. Cela se produit lorsque la paroi de l'aorte devient faible et sujette à la dilatation. Les anévrismes peuvent se former dans n'importe quelle partie de l'aorte, mais ceux qui se produisent dans l'aorte thoracique sont moins fréquents que ceux qui se produisent dans l'abdomen (anévrisme de l'aorte abdominale).
Les anévrismes de l'aorte thoracique peuvent être causés par une variété de facteurs, y compris l'athérosclérose, la maladie de Takayasu, la maladie de Marfan, la syphilis et les traumatismes. Les symptômes peuvent inclure une douleur thoracique, une toux, des difficultés respiratoires, des syncopes et des signes d'insuffisance cardiaque congestive. Cependant, de nombreux anévrismes de l'aorte thoracique ne présentent aucun symptôme et sont découverts lors d'examens d'imagerie effectués pour des raisons autres que la suspicion d'anévrisme.
Le traitement dépend de la taille, de l'emplacement et de la vitesse de croissance de l'anévrisme. Les anévrismes plus petits peuvent être surveillés par imagerie régulière, tandis que les anévrismes plus grands ou ceux qui se développent rapidement peuvent nécessiter une intervention chirurgicale pour réparer ou remplacer la section affectée de l'aorte. Le traitement médicamenteux peut également être utilisé pour gérer les facteurs de risque sous-jacents et ralentir la progression de l'anévrisme.
Le prolapsus de la valve mitrale, également connu sous le nom de maladie de Barlow ou de billowing mitral leaflet, est une affection cardiaque dans laquelle une ou les deux feuillets de la valve mitrale, situés entre les cavités supérieure et inférieure du côté gauche du cœur, sont élargis, épaissis et flasques. Cela entraîne une protrusion des feuillets dans l'atrium gauche pendant la systole, ce qui peut provoquer un reflux de sang du ventricule gauche vers l'atrium gauche (insuffisance mitrale).
Les symptômes peuvent varier d'aucun à des essoufflements, palpitations, douleurs thoraciques, fatigue et dans les cas graves, insuffisance cardiaque congestive. Le prolapsus de la valve mitrale peut être associé à d'autres affections cardiaques telles que la fibrillation auriculaire, l'endocardite infectieuse et la sténose mitrale. Il est généralement diagnostiqué par échocardiographie et peut être traité médicalement ou chirurgicalement en fonction de sa gravité et des symptômes associés.
Les chromosomes humains de la paire 15, également connus sous le nom de chromosomes 15, sont des structures en forme de bâtonnet dans les cellules du corps humain qui contiennent des gènes et de l'ADN. Chaque personne a une paire de ces chromosomes, ce qui signifie qu'il y a deux chromosomes 15 dans chaque cellule.
Les chromosomes 15 sont responsables de la régulation de diverses fonctions corporelles et du développement de certaines caractéristiques physiques. Les gènes situés sur ces chromosomes jouent un rôle important dans le fonctionnement normal du cerveau, du système immunitaire, des hormones et d'autres systèmes corporels.
Les anomalies chromosomiques de la paire 15 peuvent entraîner des troubles génétiques tels que la syndrome de l'X fragile, le syndrome de Prader-Willi et le syndrome d'Angelman. Ces conditions sont caractérisées par une variété de symptômes, notamment des retards de développement, des problèmes d'apprentissage, des troubles du comportement et des anomalies physiques.
Il est important de noter que les tests génétiques peuvent être utilisés pour détecter les anomalies chromosomiques de la paire 15 et aider à poser un diagnostic pour les personnes atteintes de ces conditions.
En termes médicaux, la généalogie est l'étude systématique des antécédents familiaux et médicaux d'une personne ou d'une famille sur plusieurs générations. Elle vise à identifier les modèles de maladies héréditaires ou génétiques dans une famille, ce qui peut aider à évaluer le risque de développer certaines affections et à mettre en œuvre des stratégies de prévention et de dépistage appropriées.
Les professionnels de la santé utilisent souvent des arbres généalogiques pour représenter visuellement les relations familiales et les antécédents médicaux. Ces outils peuvent être particulièrement utiles dans la pratique clinique, en particulier lorsqu'il s'agit de maladies rares ou complexes qui ont tendance à se produire dans certaines familles en raison de facteurs génétiques sous-jacents.
En plus d'être un outil important pour la médecine préventive, la généalogie peut également fournir des informations précieuses sur l'histoire naturelle de diverses maladies et conditions, ce qui contribue à faire progresser notre compréhension globale de la pathogenèse et de la physiopathologie. Par conséquent, elle joue un rôle crucial dans la recherche médicale et les soins cliniques.
Je suis désolé, mais la condition médicale connue sous le nom de "thorax en entonnoir" ou "thoracogenèse en entonnoir" ne semble pas être une terminologie largement reconnue ou utilisée dans la littérature médicale. Il est possible que vous ayez fait une erreur d'orthographe ou que vous vous référiez à une condition particulière qui n'est pas largement acceptée ou reconnue.
Cependant, il existe une condition appelée "pectus excavatum" qui peut donner l'apparence d'un thorax en entonnoir. Il s'agit d'une déformation congénitale de la cage thoracique où le sternum est enfoncé vers l'intérieur, ce qui donne au thorax une apparence creusée ou en forme d'entonnoir.
Si vous cherchiez une définition pour une autre condition médicale, pouvez-vous svp vérifier l'orthographe et me fournir plus de détails ? Je suis heureux de vous aider si je le peux.
L'élastine est une protéine fibreuse qui se trouve dans les tissus conjonctifs du corps humain. Elle est particulièrement concentrée dans les parois des vaisseaux sanguins, les poumons, la peau et les ligaments. L'élastine donne à ces tissus leur élasticité et leur capacité à reprendre leur forme initiale après avoir été étirés ou comprimés.
L'élastine est synthétisée par des cellules spécialisées appelées fibroblastes. Elle est composée de nombreuses chaînes polypeptidiques qui s'associent pour former des fibrilles élastiques, qui sont ensuite organisées en faisceaux plus larges pour fournir une structure solide et élastique aux tissus.
Avec l'âge, la production d'élastine peut diminuer, entraînant une perte d'élasticité des tissus et des signes de vieillissement tels que des rides cutanées et une fonction pulmonaire réduite. Des dommages à l'élastine peuvent également se produire en raison de l'exposition aux rayons UV, de la pollution atmosphérique, du tabagisme et d'autres facteurs environnementaux nocifs.
L'aorte est la plus grande artère dans le corps humain. Il s'agit d'un vaisseau musculo-élastique qui émerge du ventricule gauche du cœur et se divise en deux branches principales : l'aorte ascendante, qui monte vers le haut, et l'aorte descendante, qui descend dans la cavité thoracique et abdominale.
L'aorte a pour rôle de transporter le sang riche en oxygène des ventricules du cœur vers les différents organes et tissus du corps. Elle se ramifie en plusieurs artères plus petites qui vascularisent les différentes régions anatomiques.
L'aorte ascendante donne naissance à l'artère coronaire droite et gauche, qui approvisionnent le muscle cardiaque en sang oxygéné. L'aorte descendante se divise en deux branches : l'aorte thoracique descendante et l'aorte abdominale descendante.
L'aorte thoracique descendante donne naissance aux artères intercostales, qui vascularisent les muscles intercostaux, et à l'artère sous-clavière gauche, qui vascularise le membre supérieur gauche. L'aorte abdominale descendante se divise en plusieurs branches, dont les artères rénales, qui vascularisent les reins, et les artères iliaques, qui vascularisent les membres inférieurs.
Des maladies telles que l'athérosclérose peuvent affecter l'aorte et entraîner des complications graves, telles que la formation d'anévrismes aortiques ou la dissection aortique. Ces conditions nécessitent une prise en charge médicale et chirurgicale urgente pour prévenir les complications potentiellement fatales.
Les maladies de l'aorte se réfèrent à un large éventail de conditions qui affectent l'aorte, l'artère principale qui transporte le sang du cœur vers le reste du corps. Ces maladies peuvent inclure :
1. Aneurysme aortique : Un élargissement anormal et localisé de la paroi de l'aorte, ce qui peut entraîner une rupture ou une dissection.
2. Dissection aortique : Une condition dans laquelle le sang s'écoule entre les couches de la paroi de l'aorte en raison d'une déchirure, ce qui peut également provoquer un anévrisme ou une rupture.
3. Aortite : L'inflammation de la paroi de l'aorte, qui peut être causée par une infection, une maladie auto-immune ou d'autres facteurs.
4. Coarctation aortique : Un rétrécissement de la lumière de l'aorte, ce qui rend plus difficile pour le sang de circuler vers le bas du corps.
5. Sténose aortique : Un rétrécissement de la valve aortique, ce qui peut entraver le flux sanguin hors du cœur.
6. Hématome intramural aortique : Une accumulation de sang dans la paroi de l'aorte, souvent causée par une déchirure ou un traumatisme.
Ces maladies peuvent entraîner des complications graves, telles que des accidents vasculaires cérébraux, des crises cardiaques, des insuffisances cardiaques et la mort subite. Le traitement dépend de la gravité et du type de maladie, mais peut inclure des médicaments, des procédures endovasculaires ou des chirurgies à cœur ouvert.
Le syndrome de Down, également connu sous le nom de trisomie 21, est un trouble chromosomique causé par la présence d'une copie supplémentaire du chromosome 21. Normalement, les humains ont deux copies de chaque chromosome, un hérité de chaque parent. Le syndrome de Down se produit lorsqu'un individu a trois copies de ce chromosome, ou une partie de celui-ci, plutôt que deux.
Ce syndrome entraîne des retards de développement et des anomalies physiques caractéristiques. Les symptômes peuvent varier d'une personne à l'autre, mais ils peuvent inclure un visage plat avec une petite bouche, des oreilles basses et souvent courbées, des yeux inclinés en haut et en dehors, ainsi que des doigts courts et larges avec une unique pli cutané à la base de chaque doigt. Les personnes atteintes du syndrome de Down ont également tendance à avoir un faible tonus musculaire, des problèmes cardiaques congénitaux et un risque accru de certaines maladies infectieuses.
Le syndrome de Down est la cause la plus fréquente de retard mental et se produit dans environ une naissance sur 700. Il peut être diagnostiqué avant la naissance par des tests prénataux ou après la naissance grâce à un examen physique et à des tests chromosomiques. Actuellement, il n'existe aucun traitement pour guérir le syndrome de Down, mais des interventions éducatives, thérapeutiques et médicales peuvent aider à améliorer les capacités et la qualité de vie des personnes atteintes.
Le Syndrome Métabolique X est un terme proposé pour décrire une association de facteurs de risque cardiovasculaires et métaboliques qui vont au-delà du syndrome métabolique traditionnel. Il comprend généralement le syndrome métabolique (résistance à l'insuline, obésité abdominale, dyslipidémie et hypertension), mais il est également caractérisé par une inflammation de bas grade, une coagulation sanguine accrue, une activation du système nerveux sympathique et des perturbations du métabolisme osseux. Ce syndrome est associé à un risque élevé de développer des maladies cardiovasculaires et le diabète de type 2. Cependant, il convient de noter que la reconnaissance et la définition du Syndrome Métabolique X ne sont pas encore largement acceptées ou standardisées dans la communauté médicale.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
Une implantation de prothèse vasculaire est une procédure chirurgicale où une endoprothèse, un tube artificiel flexible, est insérée dans une artère ou une veine pour remplacer, renforcer ou élargir un vaisseau sanguin endommagé ou bloqué. Cela peut être effectué pour traiter diverses conditions telles que les anévrismes aortiques, la sténose des artères carotides, les maladies artérielles périphériques et la insuffisance veineuse.
Les endoprothèses sont généralement fabriquées à partir de matériaux tels que le Dacron (polyester) ou le PTFE (polytétrafluoroéthylène), qui sont biocompatibles et peuvent s'intégrer aux tissus environnants. Les endoprothèses peuvent être recouvertes d'un revêtement médicamenteux pour prévenir la formation de caillots sanguins ou encourager la guérison des tissus.
L'implantation de la prothèse vasculaire est généralement réalisée sous anesthésie générale ou régionale, et le chirurgien accède au vaisseau sanguin par une petite incision dans la peau. Un guidewire est inséré dans le vaisseau sanguin et utilisé pour guider l'endoprothèse à travers le vaisseau jusqu'à la zone endommagée. Une fois en place, l'endoprothèse est déployée et ajustée pour assurer une bonne adaptation à la paroi du vaisseau sanguin.
Après la procédure, le patient peut avoir besoin de prendre des médicaments anticoagulants ou antiplaquettaires pour prévenir la formation de caillots sanguins. Des contrôles réguliers peuvent également être nécessaires pour surveiller l'état de l'endoprothèse et détecter toute complication potentielle.
Le prolapsus de la valve tricuspide est une condition médicale dans laquelle la valve tricuspide, qui se trouve entre les deux dernières chambres du cœur (le ventricule droit et l'atrium droit), ne fonctionne pas correctement. Normalement, la valve tricuspide s'ouvre pour permettre au sang de circuler dans le ventricule droit pendant la relaxation cardiaque, puis se ferme hermétiquement lors de la contraction du cœur pour empêcher le reflux de sang dans l'atrium droit.
Dans le prolapsus de la valve tricuspide, une ou plusieurs des feuillets de la valve (appelés cusps) sont élargis, déformés ou affaiblis, ce qui entraîne un mauvais fonctionnement de la valve. Lorsque le cœur se contracte, les feuillets ne parviennent pas à se fermer hermétiquement, permettant ainsi au sang de refluer dans l'atrium droit. Ce reflux est également connu sous le nom de régurgitation tricuspide.
Les symptômes du prolapsus de la valve tricuspide peuvent varier considérablement, allant de presque aucun symptôme à des symptômes graves tels qu'une fatigue extrême, un essoufflement, des palpitations cardiaques et une accumulation de liquide dans les poumons (œdème pulmonaire). Dans les cas graves, le prolapsus de la valve tricuspide peut entraîner une insuffisance cardiaque droite.
Le prolapsus de la valve tricuspide est souvent associé à d'autres affections cardiaques telles que le prolapsus de la valve mitrale, les malformations congénitales du cœur et les maladies des artères coronaires. Le traitement dépend de la gravité des symptômes et peut inclure des médicaments pour soulager les symptômes, une intervention chirurgicale pour réparer ou remplacer la valve endommagée, ou une combinaison de ces deux options.
L'insuffisance aortique est un type de maladie cardiaque où la valve aortique du cœur ne parvient pas à fermer hermétiquement, permettant ainsi au sang de refluer dans l'aorte depuis la chambre de pompage principale du cœur (ventricule gauche) lorsque celui-ci se relâche. Cela peut entraîner une régurgitation ou un «fuite» du sang, ce qui affaiblit le pompage efficace du cœur et force le ventricule gauche à travailler plus fort pour maintenir un débit sanguin adéquat vers le reste de l'organisme.
Les symptômes de l'insuffisance aortique peuvent inclure des essoufflements, des palpitations, une fatigue, un gonflement des jambes et des pieds, ainsi que des douleurs thoraciques. Dans les cas graves, cette affection peut affaiblir le muscle cardiaque, ce qui conduit à une insuffisance cardiaque congestive. L'insuffisance aortique peut être causée par une maladie dégénérative de la valve aortique, une infection, un traumatisme ou une malformation congénitale. Le diagnostic est généralement posé à l'aide d'une échocardiographie, qui permet d'examiner le fonctionnement et la structure de la valve aortique. Le traitement peut inclure des médicaments pour soulager les symptômes et améliorer le fonctionnement cardiaque, mais dans certains cas graves, une intervention chirurgicale pour réparer ou remplacer la valve aortique défaillante peut être nécessaire.
L'aorte thoracique est la section de l'aorte, qui est la plus grande artère du corps humain, qui traverse la cavité thoracique. Elle s'étend du cœur à la partie supérieure de l'abdomen, où elle devient l'aorte abdominale. L'aorte thoracique assure la circulation sanguine vers les organes et les tissus du thorax, y compris les poumons, la plupart des bronches, la trachée, l'œsophage, la cage thoracique, la majorité de la colonne vertébrale et le haut de l'estomac.
Il est important de noter qu'il existe deux parties distinctes de l'aorte thoracique : l'aorte thoracique ascendante et l'aorte thoracique descendante. L'aorte thoracique ascendante s'étend du cœur à la bifurcation où elle se divise en l'aorte thoracique descendante et l'aorte brachio-céphalique, qui est le principal vaisseau sanguin desservant les bras et la tête. L'aorte thoracique descendante continue à travers la cavité thoracique jusqu'à ce qu'elle atteigne la frontière entre le thorax et l'abdomen, où elle devient l'aorte abdominale.
Les affections courantes de l'aorte thoracique comprennent l'anévrisme aortique thoracique, une dilatation localisée de la paroi aortique qui peut entraîner une rupture et des saignements catastrophiques, ainsi que la dissection aortique thoracique, dans laquelle le sang s'infiltre entre les couches de la paroi aortique, ce qui peut également provoquer une rupture. Ces conditions peuvent être causées par des maladies cardiovasculaires sous-jacentes, l'hypertension artérielle et d'autres facteurs de risque.
 Syndrome de Marfan - Wikipedia
Syndrome de Marfan - Wikipedia Syndrome de Marfan | Fondation des maladies du cœur et de l'AVC
Syndrome de Marfan | Fondation des maladies du cœur et de l'AVC Syndrome de Marfan - Pédiatrie - Édition professionnelle du Manuel MSD
Syndrome de Marfan - Pédiatrie - Édition professionnelle du Manuel MSD Santé Maghreb - Revue de presse
Santé Maghreb - Revue de presse PasseportSanté.net - Yves Cambrai (Rédacteur )
PasseportSanté.net - Yves Cambrai (Rédacteur ) Catherine Boileau : L'exploratrice de l'hypercholestérolémie familiale · Inserm, La science pour la santé
Catherine Boileau : L'exploratrice de l'hypercholestérolémie familiale · Inserm, La science pour la santé Clinique de l'aorte - Institut de cardiologie de l'Université d'Ottawa
Clinique de l'aorte - Institut de cardiologie de l'Université d'Ottawa Avertissement concernant les fluoroquinolones - ABSM asbl
Avertissement concernant les fluoroquinolones - ABSM asbl L'hyperlaxité : causes, symptômes et traitement
L'hyperlaxité : causes, symptômes et traitement Toutankhamon aurait succombé au paludisme et à une maladie des os
Toutankhamon aurait succombé au paludisme et à une maladie des os traitement Marfan - Ma Santé Blog
traitement Marfan - Ma Santé Blog myopie et astigmatisme
myopie et astigmatisme SKOPINSKI SOPHIE
SKOPINSKI SOPHIE ActuaLitté - Auteur, librairie, édition, bibliothèque : tout le livre
ActuaLitté - Auteur, librairie, édition, bibliothèque : tout le livre Adaptation d'un siège ENTHESIS pour une personne souffrant de la maladie génétique du syndrome de Malfran - Les sièges KHOL
Adaptation d'un siège ENTHESIS pour une personne souffrant de la maladie génétique du syndrome de Malfran - Les sièges KHOL Dossiers | CHL
Dossiers | CHL Fluoroquinolones
Fluoroquinolones Moxifloxacine biogaran 400 mg, comprimé pelliculé : posologie et effets secondaires | Santé Magazine
Moxifloxacine biogaran 400 mg, comprimé pelliculé : posologie et effets secondaires | Santé Magazine Forum Pseudohypoparathyroïdie : les discussions sur Carenity
Forum Pseudohypoparathyroïdie : les discussions sur Carenity Genome4Brussels: IA, Génomique & Maladies Rares - 101 Genomes Foundation
Genome4Brussels: IA, Génomique & Maladies Rares - 101 Genomes Foundation Covid-19 : L'OMS met à jour ses lignes directrices sur les traitements
Covid-19 : L'OMS met à jour ses lignes directrices sur les traitements Dystrophie musculaire | Hôpitaux Shriners pour enfants
Dystrophie musculaire | Hôpitaux Shriners pour enfants Dysphonie Spasmodique | Centre de Ressources pour les Patients Neuro - McGill University
Dysphonie Spasmodique | Centre de Ressources pour les Patients Neuro - McGill University M41.15 Scoliose juv nile idiopathique - R gion... M4115 - Code CIM 10
M41.15 Scoliose juv nile idiopathique - R gion... M4115 - Code CIM 10