Syndrome De Sturge-Weber
Tache Lie De Vin
Angiomatose
Syndrome De Klippel-Trénaunay
Malformations Maxillofaciales
Hémangiome
Méninges
Plexus Choroïde
Crises
Encéphale
Remnographie
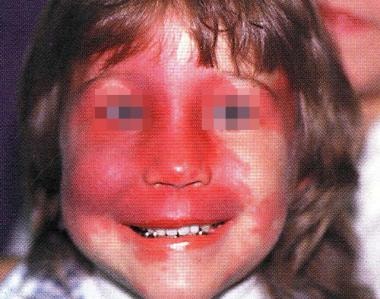
Le syndrome de Sturge-Weber est une maladie rare et congénitale qui affecte le système nerveux central et la peau. Il est également connu sous le nom de malformation angiomateuse en nappe ou d'angiome en nappe de Sturge-Weber. Cette condition est caractérisée par la présence d'un angiome plan (une tache de naissance rouge ou rose) sur le visage, souvent sur la moitié du front et du visage, qui suit le nerf trijumeau (le cinquième nerf crânien).
Ce syndrome peut également affecter le cerveau, entraînant des lésions cérébrales et une augmentation de la pression intracrânienne. Les personnes atteintes du syndrome de Sturge-Weber peuvent présenter des convulsions, des maux de tête, des troubles de la vision, des retards de développement, des problèmes d'apprentissage et des crises épileptiques. Dans certains cas, le syndrome peut également affecter les reins et les yeux.
La cause du syndrome de Sturge-Weber est inconnue, mais on pense qu'il est dû à une mutation génétique spontanée qui se produit pendant la division cellulaire très tôt dans le développement embryonnaire. Il n'existe actuellement aucun traitement curatif pour cette maladie, mais les symptômes peuvent être gérés avec des médicaments et des interventions chirurgicales.
Je suis désolé, mais la phrase que vous avez fournie, "tache lie de vin," ne semble pas être une terminologie médicale standard. Il est possible que vous fassiez référence à une "tache de vin" ou "naevus flammeus," qui est un type d'anomalie vasculaire congénitale de la peau.
Une naevus flammeus, également connu sous le nom de «marque de naissance», se caractérise par une tache plate et rose ou rouge sur la peau, qui peut varier en taille et en forme. Contrairement aux taches de vin ordinaires, qui sont généralement petites et disparaissent avec le temps, les naevus flammeus sont permanents et peuvent devenir plus foncés et plus épaisses avec l'âge.
Les naevus flammeus se produisent lorsque les vaisseaux sanguins sous la peau se développent de manière anormale pendant le développement fœtal, entraînant une accumulation de sang dans ces vaisseaux et donnant à la peau une apparence tachée. Dans la plupart des cas, les naevus flammeus ne sont pas dangereux pour la santé, mais ils peuvent être associés à certains syndromes ou troubles médicaux dans de rares cas.
Il est important de noter que toute préoccupation concernant une tache sur la peau devrait être examinée par un professionnel de la santé qualifié pour obtenir un diagnostic et des conseils appropriés.
L'angiomatose est un terme médical qui décrit une prolifération anormale de vaisseaux sanguins ou lymphatiques dans divers tissus et organes du corps. Cette affection peut affecter la peau, les muqueuses, les viscères et parfois les os.
Il existe plusieurs types d'angiomatose, en fonction de la taille et du type des vaisseaux sanguins ou lymphatiques impliqués. Les deux principaux types sont l'angiomatose vasculaire et l'angiomatose lymphatique.
L'angiomatose vasculaire est caractérisée par la présence de petits vaisseaux sanguins anormaux, appelés angiomes, qui se forment dans divers tissus et organes du corps. Les angiomes peuvent être de différentes tailles et formes, allant de petites taches cutanées à des lésions plus importantes qui peuvent affecter les organes internes.
L'angiomatose lymphatique, quant à elle, est caractérisée par la présence de vaisseaux lymphatiques anormaux qui se forment dans divers tissus et organes du corps. Ces vaisseaux lymphatiques peuvent causer une accumulation de liquide lymphatique dans les tissus, entraînant un gonflement et des lésions.
Les causes de l'angiomatose ne sont pas complètement comprises, mais il est généralement admis qu'elle résulte d'une anomalie du développement vasculaire pendant la grossesse ou dans les premiers stades de la vie. Dans certains cas, l'angiomatose peut être associée à des maladies génétiques sous-jacentes ou à une exposition à des facteurs environnementaux spécifiques.
Le traitement de l'angiomatose dépend du type et de la gravité de la maladie. Dans certains cas, aucun traitement n'est nécessaire, en particulier si les lésions sont petites et ne causent pas de symptômes. Cependant, dans d'autres cas, le traitement peut inclure des médicaments, une intervention chirurgicale ou une radiothérapie pour éliminer les lésions vasculaires anormales.
Le syndrome de Klippel-Trénaunay est un trouble congénital rare caractérisé par une combinaison de symptômes comprenant habituellement une malformation des vaisseaux sanguins (angiodysplasie), une hypertrophie des parties du corps (généralement une jambe ou un bras) et des naevus port-wine (taches de vin). Les naevus port-wine sont des taches cutanées plates, de couleur rose à pourpre, qui ne disparaissent pas au fil du temps.
Ce syndrome est généralement présent à la naissance et affecte les hommes et les femmes également. La cause exacte du syndrome de Klippel-Trénaunay n'est pas clairement comprise, mais il semble qu'il soit dû à une anomalie dans le développement des vaisseaux sanguins et lymphatiques pendant la période de développement fœtal.
Les symptômes du syndrome de Klippel-Trénaunay peuvent varier considérablement en fonction de la gravité de la maladie. Les complications courantes comprennent des douleurs chroniques, des saignements, des infections, des ulcères cutanés et des problèmes de croissance et de développement. Dans certains cas, le syndrome peut également affecter les organes internes, tels que les intestins ou les reins.
Le diagnostic du syndrome de Klippel-Trénaunay est généralement posé sur la base des antécédents médicaux et d'un examen physique complet. Des tests d'imagerie, tels que des échographies Doppler, des IRM ou des angiographies peuvent être utilisés pour confirmer le diagnostic et évaluer l'étendue de la maladie.
Le traitement du syndrome de Klippel-Trénaunay dépend de la gravité des symptômes et peut inclure une combinaison de méthodes médicales, chirurgicales et de réadaptation. Les options de traitement peuvent inclure des analgésiques pour soulager la douleur, des antibiotiques pour traiter les infections, des interventions chirurgicales pour corriger les anomalies vasculaires ou cutanées, et des thérapies de réadaptation pour aider à améliorer la fonction et la mobilité.
Les malformations maxillofaciales sont des anomalies congénitales ou acquises qui affectent la structure et la fonction des os de la mâchoire supérieure (maxillaire) et inférieure (mandibule), ainsi que des tissus environnants, y compris les dents, les muscles, les ligaments et les nerfs. Ces malformations peuvent varier en gravité, allant de légères à sévères, et peuvent affecter l'apparence physique, la fonction masticatoire, la déglutition, la parole et même la respiration.
Les exemples courants de malformations maxillofaciales comprennent :
1. Fente labiale et palatine : une ouverture dans le palais (toit de la bouche) et/ou dans les lèvres, qui peut varier en taille et en localisation.
2. Prognathisme : une mâchoire inférieure qui dépasse anormalement vers l'avant.
3. Rétrognathisme : une mâchoire inférieure reculée ou sous-développée.
4. Agénésie dentaire : l'absence congénitale de certaines dents.
5. Micrognathie : une mâchoire inférieure anormalement petite.
6. Macrogmathie : une mâchoire inférieure anormalement grande.
7. Trouble de la croissance craniofaciale : une anomalie dans le développement des os du crâne et du visage.
Le traitement des malformations maxillofaciales dépend de la gravité et de l'emplacement de la malformation, ainsi que de son impact sur la fonction et l'apparence. Les options de traitement peuvent inclure une intervention chirurgicale, une orthodontie, une thérapie de réadaptation ou une combinaison de ces approches.
Un hémangiome est un type de tumeur bénigne (non cancéreuse) qui se développe à partir des vaisseaux sanguins. Il est caractérisé par une prolifération anormale de cellules endothéliales, qui tapissent l'intérieur des vaisseaux sanguins. Les hémangiomes peuvent apparaître sur la peau ou à l'intérieur du corps, dans les organes tels que le foie, le cerveau ou les poumons.
Les hémangiomes cutanés sont les plus courants et se présentent souvent comme une petite tache rouge ou violette sur la peau, qui peut s'élargir et devenir surélevée avec le temps. Ils se développent généralement pendant les premiers mois de vie et peuvent disparaître d'eux-mêmes en quelques années.
Les hémangiomes internes peuvent ne pas provoquer de symptômes ou causer des complications dépendamment de leur taille et emplacement. Par exemple, un grand hémangiome dans le foie peut entraîner une insuffisance hépatique, tandis qu'un hémangiome cérébral peut provoquer des convulsions ou des problèmes neurologiques.
Dans la plupart des cas, les hémangiomes ne nécessitent aucun traitement car ils disparaissent spontanément. Toutefois, si un hémangiome cause des complications ou s'il est situé dans une zone esthétiquement sensible, divers traitements peuvent être envisagés, tels que la chirurgie, les lasers ou les médicaments par voie orale.
Les méninges sont des membranes protectrices qui enveloppent le cerveau et la moelle épinière. Elles sont composées de trois couches : la dure-mère, l'arachnoïde et la pie-mère. La dure-mère est la plus externe et la plus robuste, suivie de l'arachnoïde qui contient les espaces sous-arachnoïdiens remplis de liquide céphalo-rachidien. La pie-mère est la couche la plus interne et tapisse directement le cerveau et la moelle épinière. Les méninges protègent le système nerveux central contre les traumatismes physiques, assurent une barrière immunologique et maintiennent un environnement chimiquement stable pour le fonctionnement normal du cerveau et de la moelle épinière.
Le plexus choroïde est une structure anatomique du système nerveux central. Il s'agit d'un réseau complexe de vaisseaux sanguins situés dans les ventricules cérébraux, qui sont des cavités remplies de liquide à l'intérieur du cerveau. Les plexus choroïdes ont pour fonction principale de sécréter le liquide céphalo-rachidien (LCR), un fluide clair et stérile qui circule dans les ventricules et autour de la moelle épinière, protégeant ainsi le cerveau et la moelle épinière des chocs mécaniques.
Les plexus choroïdes sont formés par des capillaires sanguins recouverts d'épendymocytes, une variété de cellules épithéliales spécialisées qui régulent l'échange de nutriments, d'oxygène et de déchets entre le sang et le LCR. Les plexus choroïdes sont également impliqués dans la résorption du LCR, ce qui permet de maintenir un équilibre constant de pression et de volume du fluide céphalo-rachidien dans les ventricules cérébraux.
Des anomalies ou des malformations des plexus choroïdes peuvent être associées à certaines affections neurologiques, telles que l'hydrocéphalie, une accumulation excessive de LCR dans les ventricules cérébraux, ou des tumeurs cérébrales primitives, comme les médulloblastomes et les épendymomes. Par conséquent, une évaluation détaillée des plexus choroïdes est importante pour le diagnostic et la prise en charge de ces affections.
Un syndrome, dans le contexte médical, est un ensemble de symptômes ou de signes cliniques qui, considérés dans leur globalité, suggèrent l'existence d'une pathologie spécifique ou d'un état anormal dans le fonctionnement de l'organisme. Il s'agit essentiellement d'un ensemble de manifestations cliniques qui sont associées à une cause sous-jacente commune, qu'elle soit connue ou inconnue.
Un syndrome n'est pas une maladie en soi, mais plutôt un regroupement de signes et symptômes qui peuvent être liés à différentes affections médicales. Par exemple, le syndrome métabolique est un ensemble de facteurs de risque qui augmentent la probabilité de développer des maladies cardiovasculaires et du diabète de type 2. Ces facteurs comprennent l'obésité abdominale, l'hypertension artérielle, l'hyperglycémie à jeun et les taux élevés de triglycérides et de faibles taux de HDL-cholestérol.
La définition d'un syndrome peut évoluer avec le temps, alors que la compréhension des mécanismes sous-jacents s'améliore grâce aux recherches médicales et scientifiques. Certains syndromes peuvent être nommés d'après les professionnels de la santé qui ont contribué à leur identification ou à leur description, comme le syndrome de Down (trisomie 21) ou le syndrome de Klinefelter (XXY).
Il est important de noter que la présence d'un syndrome ne permet pas toujours d'établir un diagnostic définitif, car plusieurs affections médicales peuvent partager des symptômes similaires. Cependant, l'identification d'un syndrome peut aider les professionnels de la santé à orienter le diagnostic et le traitement vers des causes probables ou à fournir des informations sur le pronostic et la prise en charge globale du patient.
L'encéphale est la structure centrale du système nerveux situé dans la boîte crânienne. Il comprend le cerveau, le cervelet et le tronc cérébral. L'encéphale est responsable de la régulation des fonctions vitales telles que la respiration, la circulation sanguine et la température corporelle, ainsi que des fonctions supérieures telles que la pensée, la mémoire, l'émotion, le langage et la motricité volontaire. Il est protégé par les os de la boîte crânienne et recouvert de trois membranes appelées méninges. Le cerveau et le cervelet sont floating dans le liquide céphalo-rachidien, qui agit comme un coussin pour amortir les chocs et les mouvements brusques.
Une remnographie est un type d'examen d'imagerie médicale qui utilise une faible dose de radiation pour produire des images détaillées des structures internes du corps. Contrairement à une radiographie standard, une remnographie implique l'utilisation d'un milieu de contraste, comme un produit de contraste à base d'iode, qui est ingéré ou injecté dans le patient avant l'examen.
Le milieu de contraste permet aux structures internes du corps, telles que les vaisseaux sanguins, les organes creux ou les tissus mous, d'être plus visibles sur les images radiographiques. Cela peut aider les médecins à diagnostiquer une variété de conditions médicales, y compris les maladies gastro-intestinales, les maladies rénales et les troubles vasculaires.
Les remnographies sont généralement considérées comme sûres, bien que comme avec toute procédure médicale qui utilise des radiations, il existe un risque minimal de dommages aux tissus ou au matériel génétique. Les avantages potentiels d'un diagnostic précis et opportun sont généralement considérés comme dépassant ce faible risque.
Il est important de noter que les remnographies ne doivent être effectuées que lorsqu'elles sont médicalement nécessaires, car l'exposition répétée aux radiations peut augmenter le risque de dommages à long terme. Les médecins et les technologues en imagerie médicale prennent des précautions pour minimiser l'exposition aux radiations pendant les procédures de remnographie.
 Syndrome de Sturge-Weber - Wikipedia
Syndrome de Sturge-Weber - Wikipedia
 Syndrome de Sturge-Weber - Pédiatrie - Édition professionnelle du Manuel MSD
Syndrome de Sturge-Weber - Pédiatrie - Édition professionnelle du Manuel MSD
 E-CONGRES SFO
E-CONGRES SFO
 naevus; naevi
naevus; naevi
 Hétérochromie dans l'Anime - Personnages aux yeux différents
Hétérochromie dans l'Anime - Personnages aux yeux différents
 Glaucome
Glaucome
 Plaque Funéraire Personnalisée : options, matériaux et idées de design
Plaque Funéraire Personnalisée : options, matériaux et idées de design
 Montpellier
Montpellier
 Tache de vin de Porto (Nevus flammeus) chez les bébés : symptômes et causes - Bebitus
Tache de vin de Porto (Nevus flammeus) chez les bébés : symptômes et causes - Bebitus
 Elle est critiquée pour avoir enlevé au laser une tache de naissance sur le visage de son fils. "La raison n'est pas celle qu...
Elle est critiquée pour avoir enlevé au laser une tache de naissance sur le visage de son fils. "La raison n'est pas celle qu...
Angiome : Définition, risques, facteurs aggravants et traitements
 Étude de cas : hémorragie buccale après avoir mangé de la pâte d'amande
Étude de cas : hémorragie buccale après avoir mangé de la pâte d'amande
Syndrome CDG (congenital disorder of glycosylation)<...
Glossaire - Hoogstra - Centres médicaux
SYNDROME DE BAINBRIDGE-ROPERS - Page 2 - Le Forum maladies rares
COMMENT IDENTIFIER ET TRAITER LA MALADIE DE BORNHOLM - MALADIES RARES
 Maladies de l'enfant
Maladies de l'enfant
D'un4
- Syndrome de Sturge-Weber Tomodensitométrie d'un syndrome de Sturge-Weber Mise en garde médicale modifier - modifier le code - voir Wikidata (aide) Le syndrome de Sturge-Weber (ou SSW) est une maladie congénitale de la peau et du système nerveux, appartenant au groupe de phacomatoses. (wikipedia.org)
- Dans ce cas, la situation se complique car la tache de naissance peut être le signe d'un syndrome de Sturge Weber, qui peut entraîner un glaucome . (regardecettevideo.fr)
- Le syndrome de Klippel-Trenaunay associe, au niveau d'un membre, un angiome plan étendu, une augmentation de taille du membre atteint et des structures osseuses, des anomalies vasculaires plus profondes diverses. (bonne-sante.net)
- Les personnes atteintes d'un syndrome CDG ne possèdent pas l'une des enzymes nécessaires à la glycosylation. (maladiesraresinfo.org)
Glaucome2
- Dans plus de 50 % des cas, la personne touchée par un tel syndrome développera un glaucome du même côté que l'angiome plan facial de la région frontale et palpébrale supérieure, surtout si l'angiome plat (AP) occupe les paupières supérieure et inférieure. (wikipedia.org)
- Les antécédents médicaux sont un asthme bronchique et naevus flammeus congénital dans la région du front droit (syndrome de Sturge-Weber de type II) avec glaucome secondaire du côté droit. (medscape.com)
Maladie2
- Le syndrome de Sturge-Weber est une maladie vasculaire congénitale caractérisée par un angiome plan du visage de couleur lie de vin, un angiome leptoméningé et des complications neurologiques (p. ex. (msdmanuals.com)
- Les taches autour des yeux ou du front pourraient être liées à une maladie neurologique génétique rare appelée syndrome de Sturge-Weber. (bebitus.fr)
Angiome plan2
- Tout angiome plan de la région frontale et palpébrale supérieur (« tache lie-de vin ») n'est pas un syndrome de Sturge-Weber. (wikipedia.org)
- Le syndrome de Sturge-Weber-Krabbe associe un angiome plan de la moitié du front et de la paupière supérieure, des malformations des yeux et un angiome profond des méninges pouvant se traduire par des crises d'épilepsie, un retard mental et un déficit moteur. (bonne-sante.net)
N'est1
- Le syndrome de Sturge-Weber n'est pas héréditaire. (msdmanuals.com)
Tache2
- La tache est présente chez 3 bébés sur 1000 et le syndrome est près de 50 fois plus rare. (wikipedia.org)
- Le syndrome de Sturge-Weber provoque la formation d'une malformation capillaire appelé naevus lie de vin (parfois appelé tache ou marque de naissance), typiquement localisé au niveau du front et de la paupière supérieure, dans les territoires V1 et/ou V2 du nerf trijumeau. (msdmanuals.com)
D'une1
- Le type de GDC dont souffre une personne dépend de l'enzyme manquante, et les symptômes du syndrome CDG varient considérablement d'une personne à l'autre. (maladiesraresinfo.org)
20211
- Bonjour je suis la maman de Louise 2 ans edemi qui a été diagnostic en janvier 2021 du syndrome de bainbridge ropers, elle se déplace à l'aide de jeux, problèmes de sommeil elle mange des morceaux, elle ne parle pas ne nous montre pas se qu'elle veut , n'aime pas être dans les bras Trouble autistique elle a peur de la foule crise de convulsions. (maladiesraresinfo.org)
D'avoir2
- Lorsque sa topographie correspond au territoire du nerf ophtalmique, la probabilité d'avoir le syndrome est d'environ un quart. (wikipedia.org)
- Lorsque l'hétérochromie est causée par une anomalie génétique, les yeux du porteur sont plus susceptibles d'être bleus et bruns, et lorsqu'elle est causée par le syndrome, le porteur est plus susceptible d'avoir les yeux verts et bruns. (skdesu.com)
Maladies1
- Malgré le nom de syndrome ou de maladies, tous n'ont pas de réels problèmes dans la vie de la personne, mais certains doivent être pris en charge. (skdesu.com)
D'un syndrome2
- Syndrome de Sturge-Weber Tomodensitométrie d'un syndrome de Sturge-Weber Mise en garde médicale modifier - modifier le code - voir Wikidata (aide) Le syndrome de Sturge-Weber (ou SSW) est une maladie congénitale de la peau et du système nerveux, appartenant au groupe de phacomatoses. (wikipedia.org)
- Dans ce cas, la situation se complique car la tache de naissance peut être le signe d'un syndrome de Sturge Weber, qui peut entraîner un glaucome . (traveloupe.com)
Tache de naissance2
- Le syndrome de Sturge-Weber se caractérise par une tache de naissance sur le visage du nouveau-né. (msdmanuals.com)
- Les médecins suspectent un syndrome de Sturge-Weber chez les enfants qui présentent la tache de naissance caractéristique. (msdmanuals.com)
Vasculaire1
- Un autre syndrome de compression vasculaire - le syndrome de Cockett ou syndrome de May-Thurner ou syndrome de compression de la veine iliaque gauche - peut être abordé dans ce forum. (maladiesraresinfo.org)
Cerveau1
- Un syndrome neurocutané entraîne des problèmes qui affectent le cerveau, la colonne vertébrale et les nerfs (neuro), ainsi que la peau (cutané). (merckmanuals.com)
Lire1
- PMID 23656586, PMCID PMC3749068, DOI 10.1056/NEJMoa1213507, lire en ligne [html]) modifier Fiche Orphanet sur le syndrome de Sturge-Weber. (wikipedia.org)
Type1
- Cependant, ils accompagneraient également les syndromes, notamment dans les NF de type II (3 % de tous les cas). (medscape.com)