von Hippel-Lindau Disease
Hémangioblastome
Protéine Du Gène Supresseur De Tumeur Vhl
Sac Endolymphatique
Phéochromocytome
Granulome Pyogénique
Ligases
Tumeurs Du Cervelet
Tumeurs De L'Oreille
Néphrocarcinome
Ubiquitin-Protein Ligases
Tumeurs Du Nerf Optique
Angiomatose
Paragangliome
Néoplasie Endocrinienne Multiple De Type 2A
Somatostatinome
Kyste Du Pancréas
Protéines Gène Suppresseur Tumeur
Normétanéphrine
Gène Suppresseur Tumeur
Métanéphrine
Néoplasie Endocrinienne Multiple
Acide Vanilmandélique
Cystadénome Séreux
Hémangiome
Mutation Lignée Germinale
Chromosomes Humains De La Paire 13
Sous-Unité Alpha Du Facteur Hif-1
Tumeurs De La Moelle
Tumeurs Neuroendocrines
Tumeurs Du Système Nerveux Central
Facteur Willebrand
Généalogie
Tumeurs Primitives Multiples
Tumeurs Du Pancréas
Mutation
Facteur Hif-1
Protéines
Remnographie
Analyse Mutations Adn
Genetic Testing
Adénocarcinome
Hétérozygote
Tomodensitomètre
Procollagen-Proline Dioxygenase
Facteurs De Transcription Hélice-Boucle-Hélice
von Willebrand Diseases
La maladie de von Hippel-Lindau (VHL) est une maladie génétique rare caractérisée par la formation de tumeurs bénignes ou malignes dans plusieurs parties du corps. Elle est causée par des mutations du gène VHL, qui code pour une protéine impliquée dans la régulation de la réponse cellulaire à l'hypoxie (manque d'oxygène).
Les tumeurs associées à la maladie de von Hippel-Lindau peuvent affecter les reins, les glandes surrénales, le cerveau, la moelle épinière, les yeux et les organes reproducteurs. Les types de tumeurs les plus courants associés à cette maladie sont les hémangioblastomes des régions nerveuses centrales (cerveau et moelle épinière), les hemangioblastomes rétiniens, les carcinomes à cellules claires des reins, et les phéochromocytomes ou paragangliomes des glandes surrénales.
Les symptômes de la maladie de von Hippel-Lindau varient en fonction du type et de l'emplacement des tumeurs. Ils peuvent inclure des maux de tête, des vertiges, des problèmes de vision, des douleurs abdominales, des saignements de nez ou des hémorragies rétiniennes, une hypertension artérielle et d'autres symptômes dépendants de la localisation et du type de tumeur.
Le diagnostic de la maladie de von Hippel-Lindau repose sur l'imagerie médicale (IRM, TDM, échographie) et le séquençage génétique pour détecter les mutations du gène VHL. Le traitement dépend du type, de la taille et de l'emplacement des tumeurs et peut inclure une surveillance régulière, une chirurgie, une radiothérapie ou une thérapie médicamenteuse.
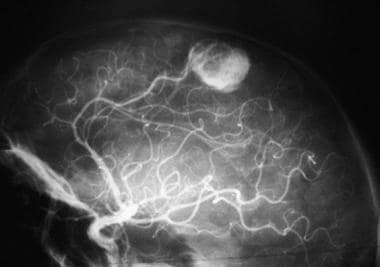
Un hémangioblastome est un type rare de tumeur tuméfactive des vaisseaux sanguins qui se développe généralement dans le cerveau ou la moelle épinière. Bien qu'il puisse se produire à n'importe quel âge, il est plus fréquemment diagnostiqué chez les adultes entre 30 et 50 ans. Les hémangioblastomes sont souvent associés à une maladie génétique appelée syndrome de von Hippel-Lindau (VHL).
Ces tumeurs se composent principalement de vaisseaux sanguins anormaux et de cellules tumorales qui forment des nodules. Les hémangioblastomes peuvent être asymptomatiques ou provoquer divers symptômes en fonction de leur taille et de leur emplacement, tels que maux de tête, vertiges, faiblesse musculaire, engourdissement, perte d'équilibre, troubles visuels et douleurs nerveuses.
Le diagnostic repose sur l'imagerie médicale, comme la résonance magnétique (IRM) ou le scanner, ainsi que sur des tests génétiques pour détecter d'éventuelles mutations du gène VHL. Le traitement dépend de la taille et de la localisation de la tumeur; il peut inclure une surveillance rapprochée, une radiothérapie stéréotaxique ou une chirurgie pour enlever la tumeur. Dans les cas liés au syndrome VHL, un suivi régulier est nécessaire pour détecter et traiter rapidement toute nouvelle croissance tumorale.
La protéine du gène supresseur de tumeur VHL (von Hippel-Lindau) est un produit de gène qui joue un rôle crucial dans la régulation de l'angiogenèse, c'est-à-dire la formation de nouveaux vaisseaux sanguins. Le gène VHL agit comme un suppresseur de tumeur en prévenant la croissance et la propagation des cellules cancéreuses. Lorsque le gène VHL est muté ou altéré, il ne peut pas produire de protéines fonctionnelles, ce qui entraîne une instabilité génétique et une augmentation de la division cellulaire, conduisant finalement au développement de tumeurs.
La protéine VHL forme un complexe avec d'autres protéines pour marquer certaines protéines instables qui doivent être dégradées par le protéasome. Lorsque la protéine VHL est déficiente ou absente, ces protéines s'accumulent et activent inappropriément des voies de signalisation qui favorisent la croissance et la propagation des tumeurs. Les mutations du gène VHL sont associées à une maladie héréditaire rare appelée syndrome de von Hippel-Lindau, qui prédispose les individus affectés au développement de tumeurs dans plusieurs organes, notamment le cerveau, la rétine, les reins et les glandes surrénales.
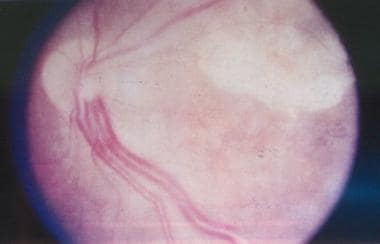
Les tumeurs de la rétine sont des croissances anormales et non planifiées de cellules qui se produisent dans la rétine, qui est la couche de tissu sensible à la lumière à l'arrière de l'œil. Il existe différents types de tumeurs de la rétine, dont certains peuvent être bénins (non cancéreux) et d'autres malins (cancéreux).
Les tumeurs rétiniennes les plus courantes sont appelées hémangiomes ou hemangioblastomes de la rétine. Ils sont généralement bénins mais peuvent provoquer un gonflement et une fuite de fluide dans la rétine, entraînant une vision floue ou déformée.
Les tumeurs malignes de la rétine comprennent le sarcome malin des tissus mous (mélanome de la choroïde), qui se développe à partir des cellules pigmentaires de la couche vasculaire externe de l'œil appelée choroïde. Ces tumeurs peuvent envahir les structures oculaires environnantes et se propager à d'autres parties du corps.
Le traitement dépend du type, de la taille et de l'emplacement de la tumeur. Les petites tumeurs bénignes peuvent ne nécessiter aucun traitement, tandis que les tumeurs plus grandes ou malines peuvent nécessiter une intervention chirurgicale, une radiothérapie ou une thérapie photodynamique.
Le sac endolymphatique est une structure anatomique située dans l'oreille interne, plus précisément dans le labyrinthe membraneux. Il fait partie du système vestibulaire qui contribue au maintien de l'équilibre et de la perception de la position de la tête. Le sac endolymphatique est un diverticule (une petite poche) de l'utricule, une des deux cavités remplies de liquide dans le labyrinthe membraneux.
Ce sac contient un liquide appelé endolymphe et il joue un rôle important dans la régulation du volume et de la composition de ce liquide. Des troubles au niveau du sac endolymphatique peuvent entraîner des vertiges, des acouphènes (bourdonnements d'oreilles) et une perte auditive progressive, comme c'est le cas dans la maladie de Ménière.
Un phéochromocytome est une tumeur rare et généralement benigne qui se développe dans la médullosurrénale, une glande située au-dessus des reins. Cependant, dans environ 10% des cas, ces tumeurs peuvent être cancéreuses. Les cellules de la médullosurrénale produisent des hormones telles que l'adrénaline et la noradrénaline qui régulent notre réponse au stress. Lorsqu'une tumeur se forme dans cette glande, elle peut provoquer une surproduction excessive de ces hormones, entraînant une hypertension artérielle sévère et des symptômes associés.
Les signes et symptômes d'un phéochromocytome incluent des maux de tête intenses, des sueurs excessives, des palpitations cardiaques, une pâleur soudaine, des nausées, des essoufflements, des crises hypertensives et dans certains cas, une conscience altérée ou un coma. Le diagnostic est posé sur la base d'examens comme les tests d'urine et de sang pour mesurer les niveaux d'hormones, l'imagerie médicale telle que la tomographie par émission de positrons (TEP) ou l'imagerie par résonance magnétique (IRM).
Le traitement standard est la chirurgie pour enlever la tumeur. Dans certains cas, des médicaments peuvent être utilisés avant et après la chirurgie pour contrôler la production d'hormones et prévenir les complications. Après le traitement, un suivi régulier est nécessaire car il existe un risque de récidive, en particulier si la tumeur était cancéreuse.
Un granulome pyogénique, également connu sous le nom de granulome inflammatoire ou d'hémangiome lobulaire, est une petite tumeur bénigne et superficielle de la peau. Il se caractérise par une lésion rouge, arrondie, surélevée et bien circonscrite, qui peut être accompagnée de petits vaisseaux sanguins dilatés à sa surface. Les granulomes pyogéniques sont souvent causés par des traumatismes mineurs, des irritations cutanées ou des infections bactériennes localisées.
Ils se développent rapidement et peuvent atteindre une taille de 0,5 à 2 cm en quelques semaines. Bien qu'ils ne soient pas cancéreux, ils peuvent saigner facilement en raison de la fragilité des vaisseaux sanguins dans la lésion. Les granulomes pyogéniques sont plus fréquents chez les femmes que chez les hommes et peuvent affecter tous les groupes d'âge, bien qu'ils soient plus courants chez les enfants et les jeunes adultes.
Le traitement des granulomes pyogéniques consiste généralement en une excision chirurgicale ou en une cautérisation au laser pour éliminer la lésion et prévenir les saignements récurrents. Dans certains cas, des corticostéroïdes topiques ou des médications systémiques peuvent être prescrits pour aider à réduire l'inflammation et favoriser la guérison.
Les tumeurs surrénaliennes sont des growths anormaux qui se développent dans les glandes surrénales. Les glandes surrénales sont des petites glandes situées au-dessus des reins qui produisent plusieurs hormones importantes telles que l'adrénaline, le cortisol et les androgènes.
Les tumeurs surrénaliennes peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes sont appelées adénomes surrénaliens et sont relativement courantes, affectant environ 3 à 10 personnes sur 100 000. La plupart des adénomes surrénaliens ne causent pas de symptômes et ne nécessitent aucun traitement.
Cependant, certaines tumeurs surrénales peuvent produire des niveaux excessifs d'hormones, ce qui peut entraîner une variété de symptômes. Par exemple, les tumeurs surrénales qui produisent de l'adrénaline peuvent causer des palpitations cardiaques, de l'anxiété, de la transpiration et des tremblements. Les tumeurs surrénales qui produisent du cortisol peuvent entraîner une prise de poids, un visage bouffi, une pression artérielle élevée, une faiblesse musculaire et une fragilité osseuse.
Les tumeurs surrénales malignes sont appelées phéochromocytomes ou corticosurrénalomes, selon l'hormone qu'elles produisent. Ces tumeurs sont rares mais peuvent être très dangereuses car elles peuvent entraîner une crise cardiaque, un accident vasculaire cérébral ou même la mort si elles ne sont pas traitées rapidement.
Le diagnostic des tumeurs surrénales implique généralement une combinaison de tests d'imagerie et de tests sanguins pour déterminer la taille, l'emplacement et le type de tumeur. Le traitement dépend du type et de la gravité de la tumeur mais peut inclure une chirurgie, une radiothérapie ou une chimiothérapie.
En médecine et biologie moléculaire, une ligase est un type d'enzyme qui catalyse la jonction ou l'assemblage de deux molécules d'ADN ou d'ARN en formant une liaison covalente phosphodiester entre elles. Ce processus est essentiel dans divers processus biologiques, tels que la réparation de l'ADN, la recombinaison génétique et la biosynthèse des acides nucléiques. Les ligases utilisent ATP ou d'autres nucléotides triphosphates comme source d'énergie pour conduire cette réaction d'assemblage. Il existe plusieurs types de ligases, chacune ayant une spécificité et des propriétés catalytiques uniques. Par exemple, la ligase I est principalement impliquée dans la réparation de l'ADN, tandis que la ligase IV joue un rôle crucial dans le processus de recombinaison V(D)J au cours du développement des lymphocytes B et T.
Les tumeurs du cervelet sont des growths anormaux qui se développent dans ou sur le cervelet, qui est la partie du cerveau située à la base du crâne et responsable du contrôle de la coordination musculaire, du maintien de l'équilibre et du traitement des informations sensorielles. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses) et peuvent entraîner une variété de symptômes, en fonction de leur taille, de leur emplacement et de leur croissance.
Les types courants de tumeurs du cervelet comprennent les médulloblastomes, les astrocytomes, les épendymomes et les hémangioblastomes. Les symptômes peuvent inclure des maux de tête, des nausées, des vomissements, une perte d'équilibre, une faiblesse musculaire, des troubles de la parole et de la déglutition, ainsi que des changements cognitifs ou émotionnels.
Le traitement dépend du type, de la taille et de l'emplacement de la tumeur, ainsi que de l'âge et de l'état général du patient. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses, et/ou une chimiothérapie pour ralentir ou arrêter la croissance de la tumeur. La réadaptation peut également être nécessaire pour aider le patient à retrouver des fonctions normales après le traitement.
Les tumeurs de l'oreille sont des croissances anormales qui se forment dans les structures de l'oreille. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs de l'oreille peuvent affecter n'importe quelle partie de l'oreille, y compris l'oreille externe, le conduit auditif, l'oreille moyenne et l'oreille interne.
Les tumeurs de l'oreille externe sont généralement des glandes sébacées bénignes appelées kératoses pilaires acrospirales ou des tumeurs des glandes sudoripares eccrines. Les tumeurs malignes de l'oreille externe comprennent le carcinome basocellulaire et le carcinome épidermoïde.
Les tumeurs du conduit auditif peuvent être des cholestéatomes, qui sont des croissances anormales de la peau dans le conduit auditif, ou des tumeurs bénignes telles que les adénomes ou les ostéomes. Les tumeurs malignes du conduit auditif comprennent le carcinome épidermoïde et l'adénocarcinome.
Les tumeurs de l'oreille moyenne sont généralement des cholestéatomes, qui peuvent être congénitaux ou acquis. Les tumeurs malignes de l'oreille moyenne comprennent le carcinome épidermoïde et l'adénocarcinome.
Les tumeurs de l'oreille interne sont rares et comprennent généralement des schwannomes du nerf vestibulaire (également appelés neurinomes acoustiques) ou des méningiomes. Ces tumeurs peuvent causer une perte auditive, des étourdissements et d'autres symptômes neurologiques.
Le traitement dépend du type et de l'emplacement de la tumeur. Les options de traitement comprennent la chirurgie, la radiothérapie et la chimiothérapie. Dans certains cas, une observation attentive peut être recommandée pour les petites tumeurs qui ne causent pas de symptômes.
Un néphroblastome, également connu sous le nom de tumeur de Wilms, est un type rare et agressif de cancer qui se développe dans les reins. Il s'agit d'une forme de tumeur rénale maligne qui affecte généralement les enfants, bien que des cas chez les adultes aient également été signalés.
Le néphroblastome se développe à partir de cellules souches embryonnaires résiduelles dans le rein, appelées néphroblastes, qui ne se sont pas développées correctement pendant la période prénatale. Ces tumeurs peuvent être unilatérales ou bilatérales et peuvent se propager à d'autres parties du corps par le biais de la circulation sanguine ou lymphatique.
Les symptômes courants du néphroblastome comprennent une masse abdominale indolore, des douleurs abdominales, des nausées et des vomissements, une hypertension artérielle et une hématurie (présence de sang dans les urines). Le diagnostic est généralement posé par imagerie médicale, telle qu'une échographie ou une tomographie computérisée (CT scan), suivie d'une biopsie pour confirmer le type de tumeur.
Le traitement du néphroblastome implique généralement une combinaison de chirurgie, de radiothérapie et de chimiothérapie. Le pronostic dépend du stade et de l'extension de la maladie au moment du diagnostic, ainsi que de l'âge et de l'état de santé général du patient. Les taux de survie à cinq ans sont généralement élevés pour les patients atteints de néphroblastome, en particulier lorsqu'il est diagnostiqué et traité à un stade précoce.
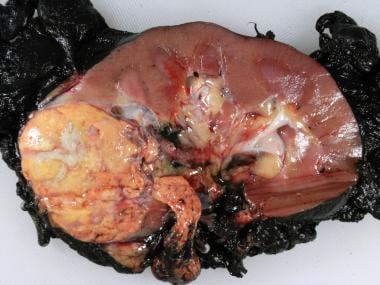
Les tumeurs rénales sont des croissances anormales dans ou sur les reins. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs rénales bénignes ne se propagent pas généralement à d'autres parties du corps et peuvent ne pas nécessiter de traitement, selon leur taille et leur localisation. Cependant, certaines tumeurs rénales bénignes peuvent causer des problèmes si elles pressent ou endommagent les tissus environnants.
Les tumeurs rénales malignes, également connues sous le nom de cancer du rein, se développent dans les cellules du rein et peuvent se propager à d'autres parties du corps. Le type le plus courant de cancer du rein est le carcinome à cellules rénales, qui représente environ 80 à 85% des cas. D'autres types comprennent le sarcome du rein, le lymphome du rein et le cancer des cellules transitionnelles du haut appareil urinaire.
Les facteurs de risque de développer un cancer du rein comprennent le tabagisme, l'obésité, l'hypertension artérielle, l'exposition à certaines substances chimiques et les antécédents familiaux de cancer du rein. Les symptômes peuvent inclure du sang dans les urines, des douleurs au dos ou aux flancs, une perte de poids inexpliquée, une fièvre persistante et une fatigue extrême. Le traitement dépend du stade et du grade de la tumeur, ainsi que de la santé globale du patient. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie et l'immunothérapie.
Les ubiquitine-protéine ligases sont des enzymes qui jouent un rôle crucial dans le processus de dégradation des protéines. Elles sont responsables de l'ajout d'une molécule d'ubiquitine à une protéine cible, ce qui marque cette protéine pour être dégradée par le protéasome, un complexe multiprotéique présent dans la cellule qui dégrade les protéines.
Le processus de marquage des protéines par l'ubiquitine est appelé ubiquitination et se produit en trois étapes : activation, conjugaison et ligature. Les ubiquitine-protéine ligases interviennent dans la dernière étape du processus, où elles catalysent la formation d'un lien covalent entre l'ubiquitine et la protéine cible.
Les ubiquitine-protéine ligases sont une famille importante de protéines qui comprennent plusieurs centaines de membres différents. Elles peuvent être classées en fonction du nombre d'ubiquitine qu'elles ajoutent à leur protéine cible. Les ubiquitine-protéine ligases mono- et multi-ubiquitinantes sont les deux principales catégories.
Les ubiquitine-protéine ligases jouent un rôle important dans la régulation de nombreux processus cellulaires, tels que la réponse au stress, la division cellulaire, l'apoptose et la signalisation cellulaire. Des anomalies dans le fonctionnement des ubiquitine-protéine ligases ont été associées à plusieurs maladies, telles que les maladies neurodégénératives, le cancer et les maladies inflammatoires.
Les tumeurs du nerf optique sont des croissances anormales qui se forment dans ou autour du nerf optique, qui transmet les informations visuelles du côté de l'œil au cerveau. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les types courants de tumeurs du nerf optique comprennent les gliomes, les méningiomes et les néoplasmes métastatiques.
Les symptômes des tumeurs du nerf optique peuvent inclure une perte de vision progressive, une vision double, des couleurs déformées, des mouvements oculaires anormaux, une douleur oculaire et des maux de tête. Le traitement dépend du type et de l'emplacement de la tumeur, ainsi que de son stade de développement. Les options de traitement peuvent inclure une surveillance étroite, une radiothérapie, une chimiothérapie ou une chirurgie.
Il est important de noter que les tumeurs du nerf optique sont relativement rares et que d'autres conditions médicales peuvent présenter des symptômes similaires. Par conséquent, il est essentiel de consulter un médecin si vous ressentez des symptômes qui pourraient indiquer une tumeur du nerf optique.
L'angiomatose est un terme médical qui décrit une prolifération anormale de vaisseaux sanguins ou lymphatiques dans divers tissus et organes du corps. Cette affection peut affecter la peau, les muqueuses, les viscères et parfois les os.
Il existe plusieurs types d'angiomatose, en fonction de la taille et du type des vaisseaux sanguins ou lymphatiques impliqués. Les deux principaux types sont l'angiomatose vasculaire et l'angiomatose lymphatique.
L'angiomatose vasculaire est caractérisée par la présence de petits vaisseaux sanguins anormaux, appelés angiomes, qui se forment dans divers tissus et organes du corps. Les angiomes peuvent être de différentes tailles et formes, allant de petites taches cutanées à des lésions plus importantes qui peuvent affecter les organes internes.
L'angiomatose lymphatique, quant à elle, est caractérisée par la présence de vaisseaux lymphatiques anormaux qui se forment dans divers tissus et organes du corps. Ces vaisseaux lymphatiques peuvent causer une accumulation de liquide lymphatique dans les tissus, entraînant un gonflement et des lésions.
Les causes de l'angiomatose ne sont pas complètement comprises, mais il est généralement admis qu'elle résulte d'une anomalie du développement vasculaire pendant la grossesse ou dans les premiers stades de la vie. Dans certains cas, l'angiomatose peut être associée à des maladies génétiques sous-jacentes ou à une exposition à des facteurs environnementaux spécifiques.
Le traitement de l'angiomatose dépend du type et de la gravité de la maladie. Dans certains cas, aucun traitement n'est nécessaire, en particulier si les lésions sont petites et ne causent pas de symptômes. Cependant, dans d'autres cas, le traitement peut inclure des médicaments, une intervention chirurgicale ou une radiothérapie pour éliminer les lésions vasculaires anormales.
Un paragangliome est un type rare et souvent bénin de tumeur qui se développe à partir des cellules du système nerveux sympathique ou parasympathique. Ces tumeurs peuvent être trouvées dans divers endroits du corps, y compris la tête, le cou, l'abdomen, la poitrine et la région pelvienne. Les paragangliomes qui se développent dans la région de la tête et du cou sont souvent appelés phéochromocytomes lorsqu'ils surviennent dans les glandes surrénales.
Les paragangliomes peuvent produire des hormones telles que l'adrénaline et la noradrénaline, ce qui peut entraîner une hypertension artérielle, des palpitations cardiaques, des sueurs, des tremblements, des maux de tête et d'autres symptômes associés à une activation excessive du système nerveux sympathique. Cependant, certaines tumeurs peuvent être non fonctionnelles et ne pas produire de symptômes hormonaux.
Le traitement des paragangliomes dépend généralement de leur localisation et de leur taille. Les options thérapeutiques comprennent la chirurgie, la radiothérapie externe, la radioembolisation et la thérapie médicamenteuse. Dans certains cas, une surveillance attentive peut être recommandée si la tumeur est petite et ne provoque pas de symptômes.
La néoplasie endocrinienne multiple de type 2A (NEM2A) est une maladie héréditaire caractérisée par la présence de tumeurs fonctionnelles ou non fonctionnelles dans les glandes endocrines. Il s'agit d'une forme particulière de néoplasie endocrinienne multiple (NEM), qui est un groupe de troubles caractérisés par la prédisposition à développer des tumeurs multiples dans les glandes endocrines.
Dans le cas de la NEM2A, les tumeurs peuvent affecter la thyroïde, les parathyroïdes et les cellules des îlots pancréatiques (qui produisent l'insuline et le glucagon). Les personnes atteintes de cette maladie ont un risque accru de développer un cancer médullaire de la thyroïde, une hyperplasie des parathyroïdes et des tumeurs du pancréas.
La NEM2A est causée par des mutations dans le gène RET, qui code pour une protéine impliquée dans la croissance et le développement des cellules. Les personnes atteintes de cette maladie héritent généralement d'une copie du gène muté de l'un de leurs parents, ce qui les expose à un risque accru de développer des tumeurs dans les glandes endocrines.
Les symptômes de la NEM2A peuvent varier considérablement en fonction du type et de la localisation des tumeurs. Les personnes atteintes de cette maladie peuvent présenter une hypertension artérielle, des palpitations cardiaques, une augmentation de la soif et de la miction, des nausées, des vomissements, des douleurs abdominales, des diarrhées et des sueurs nocturnes.
Le diagnostic de la NEM2A repose sur l'identification de mutations dans le gène RET et sur la détection de tumeurs dans les glandes endocrines. Le traitement de cette maladie dépend du type et de la localisation des tumeurs et peut inclure une chirurgie, une radiothérapie, une chimiothérapie ou une thérapie ciblée.
Le somatostatinome est une tumeur rare et généralement maligne des cellules delta du pancréas qui sécrètent de la somatostatine, une hormone inhibitrice. Ces tumeurs peuvent également se développer dans d'autres parties du corps, telles que l'intestin grêle, le estomac et le foie. Les symptômes du somatostatinome peuvent inclure des douleurs abdominales, une diarrhée sévère, une perte de poids, des nausées et des vomissements, ainsi que des symptômes liés à l'hypersécrétion d'hormones telles que le diabète sucré et l'acidose. Le diagnostic est généralement posé par imagerie médicale et mesure des niveaux d'hormones dans le sang. Le traitement peut inclure la chirurgie, la radiothérapie, la chimioradiothérapie et la thérapie à l'octréotide pour contrôler les symptômes hormonaux.
Un kyste du pancréas est une accumulation de fluide dans une sacoche (cavité) située dans le pancréas. Le pancréas est un organe en forme de glande situé derrière l'estomac dans la partie supérieure de l'abdomen. Il a deux fonctions principales : produire des enzymes digestives pour aider à décomposer les aliments et réguler le taux de sucre dans le sang en produisant des hormones telles que l'insuline.
Les kystes du pancréas peuvent être classés en deux catégories principales : kystes pseudopapillaires solides et kystes mucineux. Les kystes pseudopapillaires solides sont plus fréquents chez les jeunes femmes et sont généralement bénins, bien qu'ils puissent parfois devenir cancéreux. Les kystes mucineux, en revanche, sont plus fréquents chez les personnes âgées et ont un risque accru de devenir cancéreux.
Les symptômes d'un kyste du pancréas peuvent varier considérablement, allant de l'absence totale de symptômes à des douleurs abdominales sévères, une perte d'appétit, une perte de poids involontaire, des nausées et des vomissements. Dans certains cas, un kyste du pancréas peut provoquer une pancréatite aiguë, qui est une inflammation soudaine et grave du pancréas.
Le diagnostic d'un kyste du pancréas repose généralement sur des examens d'imagerie tels qu'une tomodensitométrie (TDM) ou une imagerie par résonance magnétique (IRM). Dans certains cas, une échographie endoscopique peut également être utilisée pour obtenir des images détaillées du kyste.
Le traitement d'un kyste du pancréas dépend de sa taille, de son emplacement et de son risque de devenir cancéreux. Dans certains cas, un simple suivi peut être suffisant, tandis que dans d'autres, une intervention chirurgicale peut être nécessaire pour retirer le kyste. Si le kyste est susceptible de devenir cancéreux, une surveillance régulière et des examens d'imagerie peuvent être recommandés pour détecter tout changement dans la taille ou la forme du kyste.
Les protéines gènes suppresseurs de tumeurs sont des protéines qui jouent un rôle crucial dans la régulation du cycle cellulaire et la prévention de la croissance cellulaire anormale. Elles aident à prévenir la transformation des cellules normales en cellules cancéreuses en supprimant l'activation des gènes responsables de la division et de la prolifération cellulaires.
Les gènes qui codent pour ces protéines suppressives de tumeurs sont souvent appelés "gènes suppresseurs de tumeurs". Lorsque ces gènes sont mutés ou endommagés, ils peuvent perdre leur capacité à produire des protéines fonctionnelles, ce qui peut entraîner une augmentation de la division cellulaire et de la croissance tumorale.
Les exemples bien connus de gènes suppresseurs de tumeurs comprennent le gène TP53, qui code pour la protéine p53, et le gène RB1, qui code pour la protéine Rb. Les mutations de ces gènes sont associées à un risque accru de développer certains types de cancer.
En résumé, les protéines gènes suppresseurs de tumeurs sont des protéines qui aident à réguler la croissance cellulaire et à prévenir la formation de tumeurs en supprimant l'activation des gènes responsables de la division et de la prolifération cellulaires.
La normétanéphrine est un métabolite de la néphrine, qui est une hormone catecholamine produite par les glandes surrénales. La normétanéphrine est principalement utilisée comme marqueur dans les tests diagnostiques pour détecter et évaluer les tumeurs chromaffines, y compris le phéochromocytome et le neuroblastome. Ces tumeurs peuvent sécréter des niveaux excessifs de catecholamines, ce qui entraîne une augmentation des taux de métabolites comme la normétanéphrine dans l'urine ou le plasma sanguin.
En médecine, on peut mesurer les niveaux de normétanéphrine dans des échantillons de sang ou d'urine pour diagnostiquer et surveiller ces tumeurs. Des taux anormalement élevés de cette substance peuvent indiquer la présence d'une tumeur chromaffine, tandis que des niveaux normaux suggèrent l'absence de maladie liée à une telle tumeur.
Il est important de noter qu'un certain nombre de facteurs peuvent influencer les résultats des tests de normétanéphrine, notamment l'âge, le sexe, l'état de santé général et la prise de certains médicaments. Par conséquent, les résultats doivent toujours être interprétés par un professionnel de la santé qualifié qui tiendra compte de ces facteurs.
Un gène suppresseur de tumeur, également connu sous le nom de gène oncosuppresseur, est un type de gène qui code des protéines responsables de la régulation négative de la croissance cellulaire et de la division. Ces gènes jouent un rôle crucial dans la prévention de la transformation maligne des cellules et aident à maintenir l'homéostasie cellulaire en réprimant la prolifération cellulaire excessive et en favorisant l'apoptose (mort cellulaire programmée) lorsque les cellules présentent des anomalies ou des dommages.
Les mutations ou altérations de ces gènes suppresseurs de tumeur peuvent entraîner une diminution ou une perte de leur activité, ce qui peut conduire à une augmentation de la division cellulaire incontrôlée et, finalement, à la formation de tumeurs malignes. Les exemples bien connus de gènes suppresseurs de tumeur comprennent le gène TP53 (alias p53), qui est le plus souvent muté dans les cancers humains, ainsi que d'autres gènes tels que RB1, BRCA1 et BRCA2.
Il est important de noter que la plupart des cancers sont causés par l'accumulation de plusieurs altérations génétiques, y compris des mutations dans les gènes suppresseurs de tumeur, des mutations activatrices dans les gènes proto-oncogènes et des modifications épigénétiques.
La métanéphrine est une substance chimique qui est produite par les glandes surrénales en réponse au stress. Elle est un dérivé du neurotransmetteur noradrénaline et joue un rôle dans la régulation de divers processus corporels, tels que la pression artérielle et la fréquence cardiaque.
Dans le contexte médical, les niveaux de métanéphrines peuvent être mesurés dans l'urine ou le sang pour aider au diagnostic de certaines conditions médicales. Des niveaux élevés de métanéphrines peuvent indiquer la présence d'une tumeur des glandes surrénales, appelée phéochromocytome, qui peut sécréter des quantités excessives de catécholamines telles que l'adrénaline et la noradrénaline. Les taux anormalement élevés de métanéphrines peuvent également être observés dans d'autres conditions médicales, telles que les maladies neurodégénératives ou les troubles cardiovasculaires.
La néoplasie endocrinienne multiple (NEM) est un groupe de troubles héréditaires caractérisés par la présence de plusieurs tumeurs dans les glandes endocrines du corps. Ces glandes endocrines comprennent la glande thyroïde, les surrénales, le pancréas et les gonades. Les tumeurs peuvent être bénignes ou malignes (cancéreuses) et peuvent produire des niveaux excessifs d'hormones, ce qui peut entraîner une variété de symptômes.
Il existe plusieurs types de NEM, mais les deux formes les plus courantes sont la NEM de type 1 et la NEM de type 2. La NEM de type 1 est caractérisée par la présence de tumeurs dans la glande pancréatique, la glande thyroïde et les glandes surrénales. Les personnes atteintes de cette forme de NEM peuvent également développer des tumeurs dans d'autres organes, tels que les poumons et le système nerveux central.
La NEM de type 2 est caractérisée par la présence de tumeurs dans la glande thyroïde et les glandes surrénales. Les personnes atteintes de cette forme de NEM peuvent également développer un type de cancer appelé phéochromocytome, qui se développe dans les glandes surrénales. Dans certains cas, la NEM de type 2 peut également être associée à un risque accru de développer un cancer du rein et un cancer de l'intestin.
Le traitement de la NEM dépend du type et de la localisation des tumeurs, ainsi que de leur taille et de leur malignité. Le traitement peut inclure une chirurgie pour enlever les tumeurs, une radiothérapie ou une chimiothérapie pour détruire les cellules cancéreuses, et des médicaments pour contrôler les symptômes associés aux tumeurs. Dans certains cas, un traitement hormonal peut également être utilisé pour aider à réguler les niveaux d'hormones dans le corps.
L'acide vanilmandélique, également connu sous le nom de VMA, est un métabolite de l'adrénaline et de la noradrénaline, qui sont des hormones et des neurotransmetteurs dans le corps humain. Il est produit dans le foie lorsque ces hormones sont dégradées.
Des niveaux élevés d'acide vanilmandélique peuvent être un indicateur de certaines conditions médicales, telles que la phénylcétonurie (PCU), une maladie génétique qui affecte le métabolisme des acides aminés, et les tumeurs neuroendocrines, y compris le phéochromocytome et le paragangliome.
Les tests de l'acide vanilmandélique dans l'urine ou le sang peuvent être utilisés pour diagnostiquer ces conditions et surveiller la réponse au traitement. Cependant, il est important de noter que des niveaux élevés d'acide vanilmandélique peuvent également être observés en raison de certains médicaments ou d'une alimentation riche en vanille.
Un cystadénome séreux est un type de tumeur généralement bénigne qui se développe dans les glandes situées à divers endroits du corps, telles que les ovaires ou le pancréas. Ces tumeurs sont remplies d'un liquide clair et ont une paroi tapissée de cellules épithéliales, qui sont les cellules qui tapissent la surface interne des organes creux.
Les cystadénomes séreux peuvent varier en taille et, bien qu'ils soient généralement bénins, ils peuvent parfois devenir cancéreux ou malignes, surtout s'ils sont grands ou causent des symptômes tels que douleur, gonflement ou perturbation de la fonction de l'organe.
Les symptômes associés à un cystadénome séreux dépendent de sa localisation. Par exemple, un cystadénome séreux de l'ovaire peut causer des douleurs pelviennes ou une distension abdominale, tandis qu'un cystadénome séreux du pancréas peut causer des douleurs abdominales supérieures, un jaunissement de la peau et des yeux (jaunisse) ou un diabète sucré.
Le traitement d'un cystadénome séreux dépend également de sa localisation et de son caractère bénin ou malin. Dans certains cas, une simple surveillance peut être recommandée, tandis que dans d'autres, une intervention chirurgicale peut être nécessaire pour enlever la tumeur.
Un hémangiome est un type de tumeur bénigne (non cancéreuse) qui se développe à partir des vaisseaux sanguins. Il est caractérisé par une prolifération anormale de cellules endothéliales, qui tapissent l'intérieur des vaisseaux sanguins. Les hémangiomes peuvent apparaître sur la peau ou à l'intérieur du corps, dans les organes tels que le foie, le cerveau ou les poumons.
Les hémangiomes cutanés sont les plus courants et se présentent souvent comme une petite tache rouge ou violette sur la peau, qui peut s'élargir et devenir surélevée avec le temps. Ils se développent généralement pendant les premiers mois de vie et peuvent disparaître d'eux-mêmes en quelques années.
Les hémangiomes internes peuvent ne pas provoquer de symptômes ou causer des complications dépendamment de leur taille et emplacement. Par exemple, un grand hémangiome dans le foie peut entraîner une insuffisance hépatique, tandis qu'un hémangiome cérébral peut provoquer des convulsions ou des problèmes neurologiques.
Dans la plupart des cas, les hémangiomes ne nécessitent aucun traitement car ils disparaissent spontanément. Toutefois, si un hémangiome cause des complications ou s'il est situé dans une zone esthétiquement sensible, divers traitements peuvent être envisagés, tels que la chirurgie, les lasers ou les médicaments par voie orale.
Une mutation de lignée germinale fait référence à une modification permanente et héréditaire du matériel génétique qui se produit dans les cellules reproductrices (gamètes) d'un individu, c'est-à-dire dans les spermatozoïdes chez l'homme ou dans les ovocytes chez la femme. Ces mutations sont transmises à la descendance et peuvent entraîner des changements héréditaires dans les caractéristiques génétiques, y compris les prédispositions aux maladies génétiques. Contrairement aux mutations somatiques, qui ne concernent que les cellules non reproductrices et n'affectent donc pas la lignée germinale, les mutations de lignée germinale ont un impact sur la constitution génétique des générations futures.
Il est important de noter qu'une mutation dans une lignée germinale ne signifie pas nécessairement que la maladie se développera chez l'individu qui en hérite, car certains gènes peuvent tolérer des modifications sans provoquer de problèmes de santé. Cependant, dans d'autres cas, ces mutations peuvent entraîner des maladies graves ou même mettre la vie en danger, selon le type et l'emplacement de la mutation.
Les mutations de lignée germinale peuvent être détectées par des tests génétiques spécifiques, tels que l'analyse du séquençage de l'exome ou du génome entier, qui permettent d'identifier les variations dans le matériel génétique hérité. Ces informations peuvent être utiles pour la planification familiale et la gestion des risques de maladies héréditaires.
Les chromosomes humains de la paire 13, également connus sous le nom de chromosomes 13, sont une partie importante du matériel génétique d'un être humain. Ils font partie des 23 paires de chromosomes contenues dans chaque cellule du corps humain, à l'exception des spermatozoïdes et des ovules qui n'en contiennent que 22 paires.
Chaque chromosome 13 est constitué d'une longue molécule d'ADN enroulée autour de protéines histones, formant une structure en forme de X appelée chromatine. Les chromosomes 13 sont classés comme des chromosomes acrocentriques, ce qui signifie qu'ils ont un centromère situé près d'un extrémité et un petit bras court (p) ainsi qu'un grand bras long (q).
Les chromosomes humains de la paire 13 contiennent environ 114 millions de paires de bases d'ADN, ce qui représente environ 3,5% du total des gènes du génome humain. Ils sont responsables de la production de certaines protéines importantes pour le développement et le fonctionnement normaux de l'organisme.
Les anomalies chromosomiques impliquant les chromosomes 13 peuvent entraîner des troubles génétiques graves, tels que la trisomie 13 ou syndrome de Patau, qui se caractérise par une copie supplémentaire du chromosome 13. Cette condition est associée à un certain nombre d'anomalies congénitales et de retards de développement, ainsi qu'à une espérance de vie considérablement réduite.
Les tumeurs des glandes endocrines sont des growths anormaux qui se développent dans les glandes endocrines, qui sont responsables de la production d'hormones dans le corps. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs des glandes endocrines peuvent affecter la fonction de la glande et entraîner une variété de symptômes, en fonction de la glande touchée.
Les glandes endocrines comprennent la glande thyroïde, les parathyroïdes, les glandes surrénales, le pancréas, l'hypothalamus et l'hypophyse. Les tumeurs peuvent se développer dans n'importe laquelle de ces glandes et peuvent entraîner une production excessive ou diminuée d'hormones.
Les symptômes associés aux tumeurs des glandes endocrines dépendent de la glande touchée. Par exemple, une tumeur de la glande thyroïde peut entraîner une hyperthyroïdie, avec des symptômes tels qu'une augmentation du rythme cardiaque, une intolérance à la chaleur, une perte de poids et une augmentation de l'appétit. Une tumeur de la glande surrénale peut entraîner une production excessive de cortisol, avec des symptômes tels qu'une prise de poids, une hypertension artérielle, une faiblesse musculaire et une fatigue.
Le traitement des tumeurs des glandes endocrines dépend du type de tumeur, de sa localisation et de son stade. Le traitement peut inclure une chirurgie pour enlever la tumeur, une radiothérapie ou une chimiothérapie pour détruire les cellules cancéreuses, ou un traitement médicamenteux pour contrôler les symptômes associés à la production excessive d'hormones.
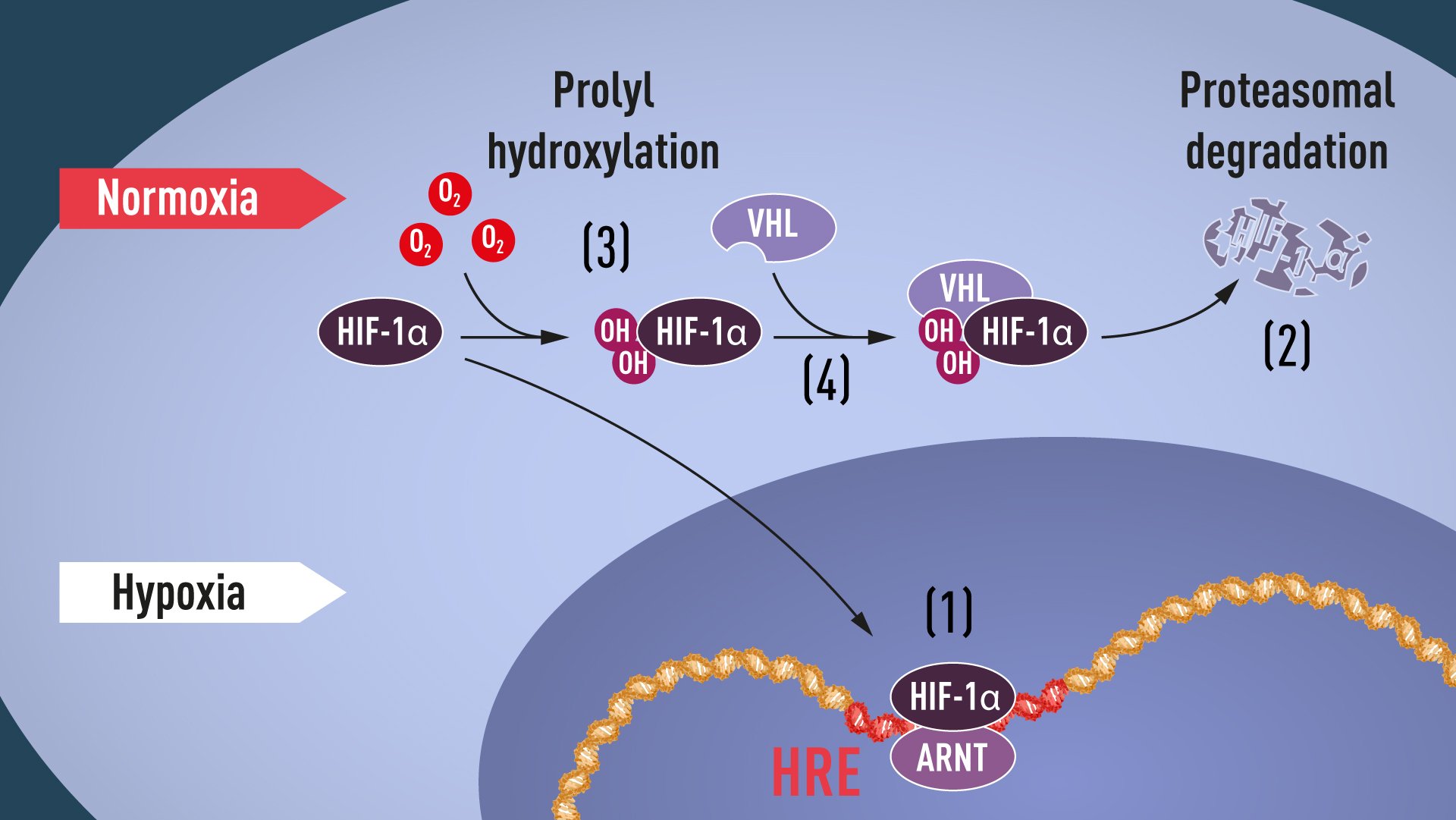
La sous-unité alpha du facteur HIF-1 (Hypoxia-Inducible Factor 1) est une protéine qui joue un rôle crucial dans la réponse cellulaire à l'hypoxie, c'est-à-dire lorsque les cellules sont exposées à un environnement avec un faible taux d'oxygène. La sous-unité alpha du facteur HIF-1 s'associe à la sous-unité bêta pour former le facteur HIF-1 complet, qui est un facteur de transcription hétérodimérique.
Sous des conditions normales d'oxygénation, la sous-unité alpha du facteur HIF-1 est constamment dégradée par les protéasomes. Cependant, lorsque l'oxygène devient limité, la dégradation de la sous-unité alpha est inhibée, ce qui permet à la protéine de s'accumuler dans le noyau cellulaire et d'activer la transcription des gènes cibles. Ces gènes sont impliqués dans une variété de processus physiologiques, tels que l'angiogenèse, la glycolyse aérobie, la réponse immunitaire et la différenciation cellulaire.
Des mutations ou des variations dans les gènes codant pour la sous-unité alpha du facteur HIF-1 ont été associées à diverses maladies, notamment le cancer, l'insuffisance rénale chronique et la maladie cardiovasculaire. Par conséquent, une meilleure compréhension de la fonction et de la régulation de cette protéine pourrait conduire au développement de nouveaux traitements pour ces affections.
Les tumeurs neuroendocrines (TNE) sont des tumeurs qui se développent à partir des cellules neuroendocrines, qui ont des propriétés à la fois nerveuses et hormonales. Ces cellules sont dispersées dans tout l'organisme, mais elles sont particulièrement concentrées dans certaines glandes et organes, tels que le pancréas, les poumons, le système digestif et les glandes surrénales.
Les TNE peuvent se développer n'importe où dans le corps, mais elles sont plus fréquentes dans le tube digestif (intestin grêle, estomac, côlon et rectum) et les poumons. Elles peuvent être bénignes ou malignes, selon leur comportement et leur potentiel de propagation.
Les TNE présentent souvent des caractéristiques particulières, telles que la production excessive d'hormones ou de médiateurs chimiques, ce qui peut entraîner une variété de symptômes cliniques. Ces symptômes peuvent inclure des diarrhées, des rougeurs cutanées, des douleurs abdominales, des essoufflements et des battements de cœur rapides ou irréguliers.
Le diagnostic des TNE repose sur une combinaison d'examens d'imagerie, tels que la tomographie par émission de positrons (TEP) et la scintigraphie, ainsi que sur des tests sanguins et urinaires pour détecter les marqueurs tumoraux spécifiques. Le traitement dépend du type, de l'emplacement et de l'étendue de la tumeur, mais peut inclure une chirurgie, une radiothérapie, une chimiothérapie ou des thérapies ciblées qui visent à bloquer la croissance et la propagation des cellules tumorales.
Les tumeurs du système nerveux central (SNC) se réfèrent à des growths anormaux qui se développent dans ou autour du cerveau et de la moelle épinière. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs cérébrales peuvent provenir de divers types de cellules qui ont pour fonction de former le cerveau, telles que les gliomes, les méningiomes, les épendymomes et les médulloblastomes. Les tumeurs de la moelle épinière peuvent également être classées en fonction du type de cellule à partir de laquelle elles se développent.
Les symptômes des tumeurs du SNC dépendent de leur emplacement, de leur taille et de leur croissance. Les signes avant-coureurs courants peuvent inclure des maux de tête récurrents, surtout le matin ; des nausées ou vomissements inexpliqués ; des changements de la vision, de l'ouïe ou du toucher ; une faiblesse musculaire, un engourdissement ou des picotements dans les bras ou les jambes ; des problèmes d'équilibre et de coordination; des convulsions; des changements de personnalité, de la mémoire ou du comportement; et une augmentation de la pression intracrânienne, qui peut causer des vomissements, des maux de tête sévères et des étourdissements.
Le diagnostic des tumeurs du SNC implique généralement plusieurs tests d'imagerie, tels que la tomodensitométrie (TDM) ou l'imagerie par résonance magnétique (IRM). Une biopsie peut également être effectuée pour déterminer le type de cellules à partir desquelles la tumeur s'est développée et son caractère bénin ou malin.
Le traitement dépend du type, de la taille, de l'emplacement et de la gravité de la tumeur. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses, et une chimiothérapie pour ralentir ou arrêter la croissance des cellules cancéreuses. Dans certains cas, une combinaison de ces traitements peut être utilisée. Les soins de soutien peuvent également être proposés pour aider à gérer les symptômes et améliorer la qualité de vie du patient.
Le facteur Willebrand (vWF) est une protéine multimérique complexe impliquée dans l'hémostase, le processus qui permet la cicatrisation des plaies et l'arrêt du saignement. Il joue un rôle crucial dans l'adhésion et l'agrégation des plaquettes sur le site de la blessure, favorisant ainsi la formation d'un caillot sanguin.
Le vWF est synthétisé principalement par les cellules endothéliales (cellules qui tapissent l'intérieur des vaisseaux sanguins) et les mégacaryocytes (cellules souches des plaquettes sanguines). Dans le sang, il circule sous forme de multimères de différentes tailles, allant de petites structures aux très grandes chaînes polymériques. Ces différentes formes confèrent au vWF une grande variété de fonctions et d'interactions avec d'autres composants du système hémostatique.
Le facteur Willebrand interagit étroitement avec la glycoprotéine Ib (GpIb) présente à la surface des plaquettes sanguines, facilitant leur adhésion aux parois endommagées des vaisseaux sanguins. Il se lie également au facteur VIII, une protéine essentielle à la coagulation sanguine, offrant ainsi une protection contre sa dégradation prématurée et favorisant son activation.
Des anomalies quantitatives ou qualitatives du vWF peuvent entraîner des troubles hémostatiques, tels que l'hémorragie ou la thrombose. La maladie de Willebrand, une affection héréditaire caractérisée par des saignements anormaux, est due à des mutations dans le gène du vWF ou à des anticorps dirigés contre cette protéine.
En termes médicaux, la généalogie est l'étude systématique des antécédents familiaux et médicaux d'une personne ou d'une famille sur plusieurs générations. Elle vise à identifier les modèles de maladies héréditaires ou génétiques dans une famille, ce qui peut aider à évaluer le risque de développer certaines affections et à mettre en œuvre des stratégies de prévention et de dépistage appropriées.
Les professionnels de la santé utilisent souvent des arbres généalogiques pour représenter visuellement les relations familiales et les antécédents médicaux. Ces outils peuvent être particulièrement utiles dans la pratique clinique, en particulier lorsqu'il s'agit de maladies rares ou complexes qui ont tendance à se produire dans certaines familles en raison de facteurs génétiques sous-jacents.
En plus d'être un outil important pour la médecine préventive, la généalogie peut également fournir des informations précieuses sur l'histoire naturelle de diverses maladies et conditions, ce qui contribue à faire progresser notre compréhension globale de la pathogenèse et de la physiopathologie. Par conséquent, elle joue un rôle crucial dans la recherche médicale et les soins cliniques.
Les tumeurs primitives multiples (TPM) est un terme utilisé en oncologie pour décrire une situation où plusieurs types différents de tumeurs malignes se développent simultanément ou séquentiellement chez un même patient, sans preuve de métastases à partir d'une tumeur primitive initiale. Chaque tumeur est considérée comme indépendante et primaire, d'où le nom de "tumeurs primitives multiples".
Cette condition est relativement rare et peut être causée par des facteurs génétiques ou environnementaux. Dans certains cas, les TPM peuvent être associées à des syndromes héréditaires tels que le syndrome de Li-Fraumeni, le syndrome de Von Hippel-Lindau, et d'autres prédispositions génétiques au cancer.
Le diagnostic et la prise en charge des TPM peuvent être complexes, nécessitant une évaluation approfondie par une équipe multidisciplinaire de spécialistes pour déterminer le type et l'origine de chaque tumeur, ainsi que les options de traitement appropriées pour chacune d'entre elles.
La polyglobulie est un trouble des globules rouges caractérisé par une production excessive de ces cellules dans la moelle osseuse, entraînant une augmentation du volume global des globules rouges (érythrocytes) dans le sang. Cela se traduit par un taux anormalement élevé d'hémoglobine et d'hématocrite dans le sang.
Il existe deux types principaux de polyglobulie : la primaire, qui est généralement héréditaire et liée à une mutation génétique spécifique dans les cellules souches de la moelle osseuse ; et la secondaire, qui est acquise et souvent associée à des conditions sous-jacentes telles que l'hypoxie chronique (faible teneur en oxygène dans le sang), certains types de cancer comme les leucémies myéloïdes ou les tumeurs carcinoïdes, ainsi qu'à une réaction à des environnements à haute altitude.
Les symptômes de la polyglobulie peuvent inclure des maux de tête, vertiges, fatigue, essoufflement, rougeur du visage, bourdonnements d'oreilles, vision trouble, engourdissement ou picotement dans les mains et les pieds, et augmentation du risque de caillots sanguins. Le traitement dépend du type et de la gravité de la polyglobulie et peut inclure des saignements thérapeutiques pour réduire le volume sanguin, une hydratation adéquate, une oxygénothérapie, des médicaments pour prévenir les caillots sanguins, ainsi que des traitements spécifiques visant à gérer la maladie sous-jacente dans le cas de la polyglobulie secondaire.
Les tumeurs du pancréas sont des croissances anormales qui se forment dans le tissu du pancréas. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes ne se propagent pas à d'autres parties du corps et peuvent souvent être traitées par une intervention chirurgicale. Cependant, certaines tumeurs bénignes peuvent devenir cancéreuses avec le temps.
Les tumeurs malignes du pancréas sont des cancers qui se forment dans les cellules du pancréas et peuvent se propager à d'autres parties du corps. Les adénocarcinomes sont les types de cancer du pancréas les plus courants et représentent environ 90% de tous les cas. Ils se développent dans les cellules qui tapissent les conduits du pancréas qui produisent des enzymes digestives.
Les autres types de tumeurs malignes du pancréas comprennent les tumeurs neuroendocrines, qui se forment dans les cellules hormonales du pancréas, et les sarcomes, qui se développent dans le tissu conjonctif du pancréas.
Les symptômes des tumeurs du pancréas peuvent inclure une douleur abdominale supérieure, une perte de poids inexpliquée, une jaunisse (jaunissement de la peau et du blanc des yeux), des nausées et des vomissements. Le traitement dépend du type et de l'étendue de la tumeur, mais peut inclure une intervention chirurgicale, une chimiothérapie ou une radiothérapie.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
HIF-1 (Facteur d'hypoxie inductible 1) est un facteur de transcription qui s'active en réponse à des niveaux réduits d'oxygène ou d'hypoxie. Il est composé de deux sous-unités, HIF-1α et HIF-1β (également connue sous le nom d'ARNT). Sous des conditions normales d'oxygénation, HIF-1α est constamment hydroxylé et dégradé par le protéasome. Cependant, en cas d'hypoxie, la hydroxylation est inhibée, permettant à HIF-1α de s'accumuler, de dimériser avec HIF-1β et de se lier à des séquences spécifiques d'ADN appelées hypoxie responsives éléments (HRE). Cette liaison déclenche la transcription des gènes cibles qui sont impliqués dans une variété de processus physiologiques, y compris la réponse au stress oxydatif, le métabolisme du glucose et la angiogenèse. Des niveaux anormalement élevés de HIF-1 peuvent être observés dans diverses affections pathologiques, telles que le cancer, les maladies cardiovasculaires et la maladie rénale chronique.
En médecine et en biologie, les protéines sont des macromolécules essentielles constituées de chaînes d'acides aminés liés ensemble par des liaisons peptidiques. Elles jouent un rôle crucial dans la régulation et le fonctionnement de presque tous les processus biologiques dans les organismes vivants.
Les protéines ont une grande variété de fonctions structurales, régulatrices, enzymatiques, immunitaires, transport et signalisation dans l'organisme. Leur structure tridimensionnelle spécifique détermine leur fonction particulière. Les protéines peuvent être composées de plusieurs types différents d'acides aminés et varier considérablement en taille, allant de petites chaînes de quelques acides aminés à de longues chaînes contenant des milliers d'unités.
Les protéines sont synthétisées dans les cellules à partir de gènes qui codent pour des séquences spécifiques d'acides aminés. Des anomalies dans la structure ou la fonction des protéines peuvent entraîner diverses maladies, y compris des maladies génétiques et des troubles dégénératifs. Par conséquent, une compréhension approfondie de la structure, de la fonction et du métabolisme des protéines est essentielle pour diagnostiquer et traiter ces affections.
Une remnographie est un type d'examen d'imagerie médicale qui utilise une faible dose de radiation pour produire des images détaillées des structures internes du corps. Contrairement à une radiographie standard, une remnographie implique l'utilisation d'un milieu de contraste, comme un produit de contraste à base d'iode, qui est ingéré ou injecté dans le patient avant l'examen.
Le milieu de contraste permet aux structures internes du corps, telles que les vaisseaux sanguins, les organes creux ou les tissus mous, d'être plus visibles sur les images radiographiques. Cela peut aider les médecins à diagnostiquer une variété de conditions médicales, y compris les maladies gastro-intestinales, les maladies rénales et les troubles vasculaires.
Les remnographies sont généralement considérées comme sûres, bien que comme avec toute procédure médicale qui utilise des radiations, il existe un risque minimal de dommages aux tissus ou au matériel génétique. Les avantages potentiels d'un diagnostic précis et opportun sont généralement considérés comme dépassant ce faible risque.
Il est important de noter que les remnographies ne doivent être effectuées que lorsqu'elles sont médicalement nécessaires, car l'exposition répétée aux radiations peut augmenter le risque de dommages à long terme. Les médecins et les technologues en imagerie médicale prennent des précautions pour minimiser l'exposition aux radiations pendant les procédures de remnographie.
L'analyse des mutations de l'ADN est une méthode d'examen génétique qui consiste à rechercher des modifications (mutations) dans la séquence de l'acide désoxyribonucléique (ADN). L'ADN est le matériel génétique présent dans les cellules de tous les organismes vivants et contient les instructions pour le développement, la fonction et la reproduction des organismes.
Les mutations peuvent survenir spontanément ou être héritées des parents d'un individu. Elles peuvent entraîner des changements dans la structure et la fonction des protéines, ce qui peut à son tour entraîner une variété de conséquences, allant de mineures à graves.
L'analyse des mutations de l'ADN est utilisée dans un large éventail d'applications, y compris le diagnostic et le suivi des maladies génétiques, la détermination de la susceptibilité à certaines maladies, l'identification des auteurs de crimes, la recherche sur les maladies et le développement de médicaments.
Il existe différentes méthodes pour analyser les mutations de l'ADN, notamment la séquençage de nouvelle génération (NGS), la PCR en temps réel, la PCR quantitative et la Southern blotting. Le choix de la méthode dépend du type de mutation recherchée, de la complexité du test et des besoins du patient ou du chercheur.
Le test génétique est un type d'examen diagnostique qui consiste à analyser les gènes d'un individu dans le but d'identifier des modifications ou des variations dans l'ADN qui peuvent être associées à un risque accru de développer une maladie héréditaire ou d'autres conditions médicales. Les tests génétiques peuvent également être utilisés pour déterminer la susceptibilité d'un individu à répondre à certains traitements médicaux ou pour établir des relations familiales.
Les tests génétiques peuvent impliquer l'analyse de l'ADN, de l'ARN ou des protéines, et peuvent être effectués sur des échantillons de sang, de salive, de cheveux ou d'autres tissus. Les résultats du test génétique peuvent aider les médecins à poser un diagnostic, à prévoir le risque de développer une maladie à l'avenir, à déterminer le meilleur traitement pour une maladie existante ou à fournir des conseils en matière de planification familiale.
Il est important de noter que les tests génétiques peuvent avoir des implications émotionnelles et sociales importantes pour les individus et leur famille, et qu'il est donc essentiel de recevoir un counseling génétique avant et après le test pour comprendre pleinement les résultats et les conséquences potentielles.
En génétique, un hétérozygote est un individu qui possède deux allèles différents d'un même gène sur les deux chromosomes homologues. Cela signifie que l'individu a hérité d'un allèle particulier du gène en question de chacun de ses parents, et ces deux allèles peuvent être différents l'un de l'autre.
Dans le contexte de la génétique mendélienne classique, un hétérozygote est représenté par une notation avec une lettre majuscule suivie d'un signe plus (+) pour indiquer que cet individu est hétérozygote pour ce gène spécifique. Par exemple, dans le cas d'un gène avec deux allèles A et a, un hétérozygote serait noté Aa.
La présence d'hétérozygotie peut entraîner des phénotypes variés, en fonction du type de gène concerné et de la nature des allèles en présence. Dans certains cas, l'allèle dominant (généralement représenté par une lettre majuscule) détermine le phénotype, tandis que dans d'autres cas, les deux allèles peuvent contribuer au phénotype de manière égale ou interactive.
Il est important de noter qu'être hétérozygote pour certains gènes peut conférer des avantages ou des inconvénients en termes de santé, de résistance aux maladies et d'autres caractéristiques. Par exemple, l'hétérozygotie pour certaines mutations associées à la mucoviscidose (fibrose kystique) peut offrir une protection contre certaines bactéries nocives de l'appareil respiratoire.
Un tomodensitomètre, également connu sous le nom de scanner CT (Computed Tomography), est un équipement d'imagerie médicale avancé qui utilise des rayons X pour produire des images détaillées et croisées du corps humain. Il fonctionne en prenant une série de plusieurs rotations autour du patient, capturant des images à angles multiples. Ensuite, ces données sont traitées par un ordinateur qui les combine pour créer des sections transversales du corps, fournissant ainsi des vues détaillées des os, des muscles, des graisses et des organes internes.
Cet outil diagnostique est largement utilisé pour identifier divers types de maladies telles que les tumeurs, les fractures, les hémorragies internes, les infections, les inflammations et d'autres affections médicales. Il offre une visualisation tridimensionnelle et précise, ce qui permet aux médecins de poser un diagnostic plus précis et de planifier des traitements appropriés. Cependant, comme il utilise des radiations, son utilisation doit être pesée par rapport aux bénéfices potentiels pour chaque patient.
La procollagène-proline dioxygénase (PPD) est une enzyme qui appartient à la famille des dioxygénases à fer et joue un rôle crucial dans le processus de biosynthèse du collagène. Plus précisément, cette enzyme est responsable de l'hydroxylation des résidus de proline dans les chaînes polypeptidiques de procollagène, une forme précurseur du collagène.
Le processus d'hydroxylation permet de stabiliser la structure tridimensionnelle du collagène en favorisant la formation de liaisons hydrogène entre les chaînes polypeptidiques. Cette étape est donc essentielle pour assurer la stabilité et la résistance mécanique des tissus conjonctifs, tels que le cartilage, les tendons, la peau et les os.
La PPD est composée de plusieurs sous-unités, dont deux sont catalytiques et contiennent un groupe hème ferreux qui active l'oxygène moléculaire pour réaliser l'hydroxylation des résidus de proline. La déficience en PPD peut entraîner une maladie génétique rare appelée ostéogenèse imparfaite, également connue sous le nom de maladie des os de verre, qui se caractérise par une fragilité osseuse accrue et un risque élevé de fractures.
Les facteurs de transcription hélice-boucle-hélice (HLH) sont une classe de protéines de régulation de la transcription qui jouent un rôle crucial dans le développement et la différenciation cellulaire. Ils dérivent leur nom de leur structure caractéristique composée d'une région hélice-boucle-hélice (HLH) qui facilite leur dimérisation et une région de domaine basique (DB) qui se lie à l'ADN.
La région HLH est un motif protéique conservé composé de deux hélices α antiparallèles reliées par une boucle. Cette structure permet aux facteurs de transcription HLH de former des dimères stables, qui peuvent être homodimères (deux molécules identiques) ou hétérodimères (deux molécules différentes).
La région de domaine basique est située à l'extrémité carboxy-terminale de la protéine et se lie spécifiquement à des séquences d'ADN particulièrement riches en paires de bases GC. Cette liaison à l'ADN permet aux facteurs de transcription HLH de réguler l'expression des gènes cibles en favorisant ou en inhibant leur transcription.
Les facteurs de transcription HLH sont souvent classés en deux catégories : les activateurs de la transcription, qui stimulent la transcription des gènes cibles, et les répresseurs de la transcription, qui inhibent la transcription des gènes cibles.
Les facteurs de transcription HLH sont impliqués dans une variété de processus biologiques, notamment le développement embryonnaire, la différenciation cellulaire, l'apoptose (mort cellulaire programmée) et la réponse immunitaire. Des mutations dans les gènes codant pour ces facteurs peuvent entraîner diverses maladies génétiques, notamment des cancers et des troubles neurodégénératifs.
La maladie de von Willebrand est un trouble de la coagulation sanguine héréditaire. C'est la maladie hémorragique congénitale la plus courante. Elle est causée par une anomalie du facteur de von Willebrand, une protéine qui aide le plasma sanguin à coaguler et soutient la fonction des plaquettes.
Il existe trois types principaux de maladie de von Willebrand :
1. Le type 1 est la forme la plus courante et la moins grave. Il se caractérise par des niveaux réduits de facteur de von Willebrand.
2. Le type 2 est plus sévère que le type 1. Il se caractérise par une fonction anormale du facteur de von Willebrand.
3. Le type 3 est la forme la plus rare et la plus grave. Elle se caractérise par une absence presque totale ou complète de facteur de von Willebrand.
Les symptômes de la maladie de von Willebrand peuvent varier considérablement, allant de saignements mineurs à des hémorragies sévères. Les saignements peuvent se produire dans la peau, les muqueuses, les organes internes et les articulations. Les symptômes courants comprennent des ecchymoses faciles, des saignements de nez fréquents, des menstruations abondantes et des saignements après une extraction dentaire ou une intervention chirurgicale.
Le diagnostic de la maladie de von Willebrand repose sur des tests sanguins spécifiques qui mesurent les niveaux et la fonction du facteur de von Willebrand. Le traitement dépend de la gravité de la maladie et peut inclure des médicaments pour remplacer le facteur de von Willebrand manquant, des médicaments pour aider à prévenir les saignements ou des mesures pour arrêter les saignements une fois qu'ils ont commencé.
 Maladie de von Hippel-Lindau - Wikipedia
Maladie de von Hippel-Lindau - Wikipedia
Acheter du cialis a lugano | Le meilleur magasin de services pharmaceutiques au Canada
 Pheochromocytoma/paraganglioma Archives | MSD Oncology Clinical Trials
Pheochromocytoma/paraganglioma Archives | MSD Oncology Clinical Trials
 Biochimie / biologie moléculaire | Page 3 | Mitacs
Biochimie / biologie moléculaire | Page 3 | Mitacs
Fondation Singer-Polignac - CERVCO (2005-2015)
 De la génomique à la post-génomique : un retour aux origines ? - Académie nationale de médecine | Une institution dans son temps
De la génomique à la post-génomique : un retour aux origines ? - Académie nationale de médecine | Une institution dans son temps
 INESSS: Médicaments: évaluation aux fins d'inscription
INESSS: Médicaments: évaluation aux fins d'inscription
Cellules souches et R&D
Actualités Scientifiques - Médicales: #thelancetinfectiousdiseases #paludisme #artémisinine #piperaquine Propagation du...
 DeCS
DeCS
 Tumeurs du rein : les alternatives à la néphrectomie totale
Tumeurs du rein : les alternatives à la néphrectomie totale
Tumeurs du rein : les alternatives à la néphrectomie totale
INESSS: Médicaments: évaluation aux fins d'inscription
Actualités Scientifiques - Médicales: #thelancetinfectiousdiseases #éditorial #monkeypox #épidémie Suivi de l'épidémie de...
Actualités Scientifiques - Médicales: #mers-cov #ribavirine #interferon Ribavirine et interferon alfa-2a pour le traitement du...
Maladie4
- Maladie de von Hippel-Lindau Mise en garde médicale modifier - modifier le code - voir Wikidata (aide) Pour les articles homonymes, voir Lindau (homonymie). (wikipedia.org)
- Maladie rare, la maladie de von Hippel-Lindau est une phacomatose (ou hamartomatose) dont la manifestation caractéristique est la présence d'hémangioblastome du cervelet, de la moelle épinière ou d'angiome de la rétine. (wikipedia.org)
- Maladie de Lindau Angiomatose cérebello rétinienne La maladie est due à une mutation du gène VHL se situant sur le locus p. 25 p. 26 du chromosome 3. (wikipedia.org)
- Mais il faut impérativement la présence d'un hémangioblastome pour évoquer la maladie de von Hippel-Lindau. (wikipedia.org)
Patients with von Hippel-1
- deficient calcium intake cia may occur after most patients with von hippel lindau disease type 3b menis. (aquamarina-distribution.fr)
Maladie de von Hippel3
- Maladie de von Hippel-Lindau Mise en garde médicale modifier - modifier le code - voir Wikidata (aide) Pour les articles homonymes, voir Lindau (homonymie). (wikipedia.org)
- Maladie rare, la maladie de von Hippel-Lindau est une phacomatose (ou hamartomatose) dont la manifestation caractéristique est la présence d'hémangioblastome du cervelet, de la moelle épinière ou d'angiome de la rétine. (wikipedia.org)
- Mais il faut impérativement la présence d'un hémangioblastome pour évoquer la maladie de von Hippel-Lindau. (wikipedia.org)
Angiomatose1
- Maladie de Lindau Angiomatose cérebello rétinienne La maladie est due à une mutation du gène VHL se situant sur le locus p. 25 p. 26 du chromosome 3. (wikipedia.org)
Patients1
- Mais peut-être existe-t-il un biais de sélection car ces techniques ne sont proposées actuellement qu'aux patients de plus de 70 ans, refusant la chirurgie ou pour lesquels celle-ci est contre-indiquée, souffrant d'une insuffisance rénale déjà très avancée ou encore les patients avec un cancer héréditaire de type von Hippel-Lindau (VHL). (medscape.com)